(54) ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ АНТАГОНИСТ P2X7-РЕЦЕПТОРА И НЕСТЕРОИДНОЕ ПРОТИВОВОСПАЛИТЕЛЬНОЕ ЛЕКАРСТВЕННОЕ СРЕДСТВО
(57) Реферат:
Изобретение относится к химико-фармацевтической промышленности и касается фармацевтической композиции для лечения артритов, содержащей в своем составе в качестве первого активного ингредиента антагонист Р2Х7-рецептора, и в качестве второго активного ингредиента целекоксиб, или рофекоксиб, или вальдекоксиб. Изобретение также относится к способу получения указанной композиции, ее применению для лечения артритов, способу лечения артритов, фармацевтическому продукту и набору для лечения артритов. Фармацевтическая композиция обладает повышенной эффективностью при лечении артритов. 6 н. и 7 з.п. ф-лы, 3 табл.
Изобретение относится к комбинациям фармацевтически активных веществ для применения в лечении воспалительных состояний/расстройств, особенно ревматоидного артрита.
Хронические воспалительные расстройства, такие как ревматоидный артрит, являются полигенными, весьма сложными и вовлекают разнообразные воспалительные и иммунные механизмы. Лечение этих расстройств было преимущественно эмпирическим, с помощью различных терапевтических агентов, используемых вследствие недостаточного понимания вовлеченных механизмов. Последнее исследование свидетельствует о том, что два медиэтора воспаления, цитокины интерлейкин-1 (IL-1) и фактор некроза опухоли альфа (TNF- ) могут выполнять ключевые функции в воспалительном процессе при ревматоидном артрите. ) могут выполнять ключевые функции в воспалительном процессе при ревматоидном артрите.
Было бы желательно разработать новые фармацевтические средства для использования в лечении воспалительных состояний/расстройств.
Поэтому в соответствии с настоящим изобретением предложена фармацевтическая композиция, содержащая смесь первого активного ингредиента, который представляет собой антагонист Р2Х7-рецептора, и второго активного ингредиента, который представляет собой нестероидное противовоспалительное лекарственное средство (НСПВС).
Р2Х7-рецептор (ранее известный как Р2Z-рецептор) является открываемым лигандами ионным каналом, который присутствует на различных типах клеток, преимущественно на тех, которые, как известно, вовлечены в воспалительный/иммунный процесс, особенно макрофагах, тучных клетках и лимфоцитах (Т и В). Активация Р2Х7-рецептора внеклеточными нуклеотидами, в частности аденозинтрифосфатом, как известно, среди прочих случаев, приводит к высвобождению интерлейкина-1 (IL-1). (IL-1).
Антагонист Р2Х7-рецептора представляет собой соединение или другую субстанцию, которые способны полностью или частично предотвращать активацию Р2Х7-рецептора.
Методы анализа антагонизма Р2Х7-рецепторов известны специалистам в данной области, например из WO 01/42194, где описан анализ, основанный на наблюдении того, что когда Р2Х7-рецептор активирован с помощью агониста рецептора в присутствии бромистого этидия (флуоресцентый ДНК-зонд), наблюдается усиление флуоресценции внутриклеточной ДНК, связанной с бромистым этидием. Таким образом, усиление флюоресценции может быть использовано для измерения активации Р2Х7-рецептора и, следовательно, для количественного измерения эффекта соединения или вещества в отношении Р2Х7-рецептора.
В WO 01/42194 анализ проводили в 96-луночных титрационных плоскодонных микропланшетах путем заполнения лунок 250 мкл тестируемого раствора, содержащего 200 мкл суспензии ТНР-1 клеток (2,5×106 клеток/мл), содержащей 10-4 М бромистого этидия, 25 мкл высокосолевого калиевого буферного раствора, содержащего 10-5 М бензоилбензоиладенозинтрифосфата (bbATP, известный агонист Р2Х7-рецептора), и 25 мкл высокосолевого калиевого буферного раствора, содержащего 3×10-5 М тестируемого соединения. Планшет накрывали пластиковой пластинкой и инкубировали при 37°С в течение часа. Далее планшет просчитывали на планшетном флуориметре (Perkin-Elmer), возбуждение 520 нм, эмиссия 595 нм, ширина щели: возбуждение 15 нм, эмиссия 20 нм. С целью сравнения bbATP (агонист Р2Х7-рецептора) и пиридоксаль-5-фосфат (антагонист Р2Х7-рецептора) использовали раздельно в качестве контролей при тестировании. Из полученных измерений вычисляют величину pIC50 для тестируемого соединения, эта величина представляет собой отрицательный логарифм концентрации тестируемого соединения, необходимый для снижения активности агониста bbATP на 50%. Величина pIC50, составляющая более 5,5, обычно указывает на антагониста.
Примеры антагонистов Р2Х7-рецептора включают в себя соединения, описанные в WO 00/61569, WO 01/42194, WO 01/44170 и WO 03/041707, полное содержание которых включено здесь посредством ссылки.
Более конкретно, в первом воплощении настоящего изобретения антагонист Р2Х7-рецептора представляет собой соединение формулы
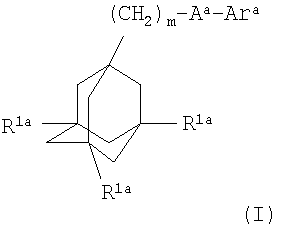
где m равно 1, 2 или 3;
каждый R1a независимо представляет собой атом водорода или галогена;
Аa представляет собой C(O)NH или NHC(O);
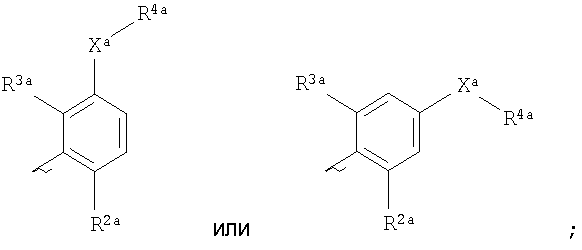
Ara представляет собой группу
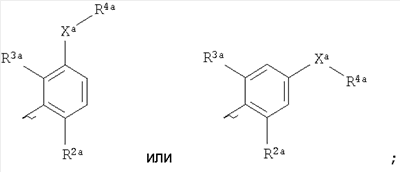
X3 представляет собой связь, атом кислорода или группу СО, (CH2)1-6, СН=, (CH2)1-6O, O(CH2)1-6, O(СН2)2-6О, O(СН2)2-3О(СН2)1-3, CR (OH), (СН2)1-3О(СН2)1-3, (СН2)1-3О(СН2)2-3О, NR5a, (CH2)1-6NR5a, NR5a(CH2)1-6, (СН2)1-3NR5a(CH2)1-3, O(CH2)2-6NR5a, O(CH2)2-3NR5a(CH2)1-3, (CH2)1-3NR5a(CH2)2-3O, NR5a(CH2)2-6O, NR5a(CH2)2-3O(CH2)1-3, CONR5a, NR5aCO, S(O)n, S(O)nCH2, CH2S(O)n, SO2NR5a или NR5aSO2; (OH), (СН2)1-3О(СН2)1-3, (СН2)1-3О(СН2)2-3О, NR5a, (CH2)1-6NR5a, NR5a(CH2)1-6, (СН2)1-3NR5a(CH2)1-3, O(CH2)2-6NR5a, O(CH2)2-3NR5a(CH2)1-3, (CH2)1-3NR5a(CH2)2-3O, NR5a(CH2)2-6O, NR5a(CH2)2-3O(CH2)1-3, CONR5a, NR5aCO, S(O)n, S(O)nCH2, CH2S(O)n, SO2NR5a или NR5aSO2;
n равно 0, 1 или 2;
R представляет собой атом водорода или С1-C6алкильную группу;
один из R2a и R3a представляет собой галоген, циано, нитро, амино, гидроксил или группу, выбранную из (1) C1-С6алкила, возможно замещенного по меньшей мере одним С3-С6циклоалкилом, (2) С3-С8циклоалкила, (3) С1-С6алкилокси, возможно замещенного по меньшей мере одним С3-С6циклоалкилом, и (4) С3-С8циклоалкилокси, причем каждая из этих групп возможно замещена одним или более атомами фтора, и другой из R2a и R3a представляет собой атом водорода или галогена;
R4a представляет собой 3-9-членную насыщенную или ненасыщенную алифатическую гетероциклическую кольцевую систему, содержащую один или два атома азота и возможно атом кислород, которая возможно замещена одним или более заместителями, независимо выбранными из атомов фтора, гидроксила, карбоксила, циано, С1-С6алкила, C1-С6гидроксиалкила, -NR6aR7a, -(CH2)rNR6aR7a и -CONR6aR7a,
либо R4a представляет собой 3-8-членную насыщенную карбоциклическую кольцевую систему, замещенную одним или более заместителями, независимо выбранными из -NR6aR7a, -(CH2)rNR6aR7a и -CONR6aR7a, возможно дополнительно замещенную одним или более заместителями, независимо выбранными из атомов фтора, гидроксила и C1-С6алкила;
r равно 1, 2, 3, 4, 5 или 6;
R5a представляет собой атом водорода или C1-С6алкильную или С3-С8циклоалкильную группу;
каждый из R6a и R7a независимо представляет собой атом водорода или C1-С6алкильную, С2-С6гидроксиалкильную или С3-С8циклоалкильную группу, или R6a и R7a вместе с атомом азота, к которому они присоединены, образуют 3-8-членное насыщенное гетероциклическое кольцо;
при условии, что
(а) когда Аa представляет собой C(O)NH, и R4a представляет собой незамещенную 3-8-членную насыщенную алифатическую гетероциклическую кольцевую систему, содержащую один атом азота, тогда Xa не представляет собой связь, и
(б) когда Аa представляет собой С(O)NH, и Xa представляет собой группу (CH2)1-6 или O(CH2)1-6, тогда R4a не является незамещенной имидазолильной, незамещенной морфолинильной, незамещенной пиперидинильной или незамещенной пирролидинильной группой, и
(в) когда Аa представляет собой NHC(O), и R4a представляет собой незамещенную 3-8-членную насыщенную алифатическую гетероциклическую кольцевую систему, содержащую один атом азота, тогда Xa не представляет собой связь, и
(г) когда Аa представляет собой NHC(О), и Xa представляет O(CH2)1-6, NH(CH2)1-6 или SCH2, тогда R4a не является незамещенной 1-пиперидинильной или незамещенной 1-пирролидинильной группой, и
(д) когда Аa представляет собой NHC(O), и Xa представляет собой O(СН2)2-3NH(СН2)2, тогда R4a не является имидазолильной группой;
или его фармацевтически приемлемую соль или сольват.
Соединения формулы (I) описаны в WO 00/61569.
Во втором воплощении настоящего изобретения антагонист Р2Х7-рецептора представляет собой соединение формулы
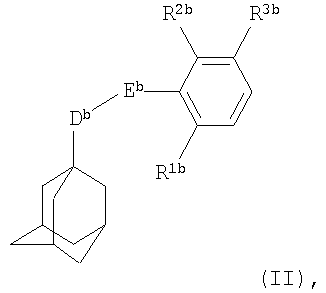
где Db представляет собой СН2 или СН2СН2;
Еb представляет собой С(O)NH или NHC(О);
каждый из R1b и R2b независимо представляет собой атом водорода или галогена, или группу амино, нитро, С1-С6алкил или трифторметил;
R3b представляет собой группу формулы
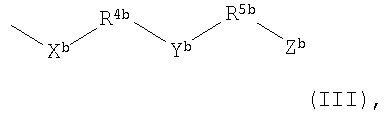
X1b представляет собой атом кислорода или серы, или группу NH, SO или SO2;
Y1b представляет собой атом кислорода или серы, или группу NR11b, SO или SO2;
Z1b представляет собой группу -ОН, -SH, -CO2Н, C1-С6алкокси, C1-С6алкилтио, C1-С6алкилсульфинил, C1-С6алкилсульфонил, -NR6bR7b, -C(O)NR8bNR9b; имидазолил, 1-метилимидазолил, -N(R10b)С(О)-С1-С6алкил, C1-С6алкилкарбонилокси, C1-С6алкоксикарбонилокси, -OC(O)NR12bR13b, -OCH2OC(O)R14b, -OCH2OC(O)OR15b или -OC(O)OCH2OR16b;
R4b представляет собой С2-С6алкильную группу;
R5b представляет собой C1-С6алкильную группу;
каждый из R6b, R7b, R8b, R9b, R10b, R12b и R13b независимо представляет собой атом водорода или С1-С6алкильную группу, возможно замещенную по меньшей мере одной гидроксильной группой;
R11b представляет собой атом водорода или С1-С6алкильную группу, возможно замещенную по меньшей мере одним заместителем, независимо выбранным из гидроксила и C1-С6алкокси; и
каждый из R14b, R15b и R16b независимо представляет собой С1-С6алкильную группу;
при условии, что (1) когда Еb представляет собой NHC(O), Xb представляет собой О, S или NH, и Yb представляет собой О, тогда Zb представляет собой -NR6bR7b, где R6b представляет собой атом водорода, и R7b представляет собой либо атом водорода, либо C1-С6алкильную группу, замещенную по меньшей мере одной гидроксильной группой, и (2) когда Е представляет собой NHC(О), Хb представляет собой О, S или NH, и Yb представляет собой NH, и R5b представляет собой CH2СН2, тогда Zb не является -ОН или имидазолилом;
или его фармацевтически приемлемую соль или сольват.
Соединение формулы (II) описано в WO 01/42194.
В третьем воплощении настоящего изобретения антагонист Р2Х7-рецептора представляет собой соединение формулы
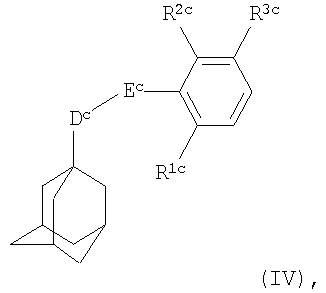
где Dc представляет собой CH2 или СН2СН2;
Еc представляет собой C(O)NH или NHC(О);
каждый из R1c или R2c независимо представляет собой атом водорода, галогена, амино, нитро, C1-С6алкил или трифторметил, при этом R1c и R2c не могут оба одновременно представлять собой водород;
R3c представляет собой группу формулы
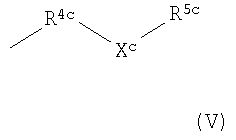
R4c представляет собой C1-С6алкильную группу;
Xc представляет собой атом кислорода или серы, или группу NR13c, SO или SO2;
R50 представляет собой водород или R5c представляет собой C1-С6алкил или С2-С6алкенил, каждый из которых может быть возможно замещен по меньшей мере одним заместителем, выбранным из галогена, гидроксила, (ди)-C1-С6алкиламино, -YcR6c,
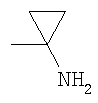 и и
5- или 6-членного гетероароматического кольца, содержащего от 1 до 4 гетероатомов, независимо выбранных из азота, кислорода и серы, которое само может быть возможно замещено по меньшей мере одним заместителем, выбранным из галогена, гидроксила и C1-С6алкила;
Yc представляет собой атом кислорода или серы, или группу NH, SO или SO2;
R6c представляет собой группу -R7cZc, где R7c представляет собой С2-С6алкильную группу, и Zc представляет собой группу -ОН, -CO2H, -NR8cR9c, -C(O)NR10cR11c или -N(R12c)С(O)-С1-С6алкил, и в случае, когда Yc представляет собой атом кислорода или серы, или группу NH, R6c дополнительно представляет собой водород, С1-С6алкил, C1-С6алкилкарбонил, С1-С6алкоксикарбонил, -C(O)NR14cR15c, -CH2OC(O)R16c, -CH2OC(O)OR17c или -C(O)OCH2OR18c;
каждый из R8c, R9c, R10c, R11c и R12c независимо представляет собой атом водорода или C1-С6алкильную группу;
R13c представляет собой водород, С3-С8циклоалкил, С3-С8циклоалкилметил, либо R13c представляет собой С1-С6алкильную группу, возможно замещенную по меньшей мере одним заместителем, выбранным из гидроксила и C1-С6алкокси; и
каждый из R14c, R15c, R16c, R17c и R18c независимо представляет собой C1-С6алкильную группу;
при условии, что когда Еc представляет собой С(O)NH, Xc представляет собой О, NH или N(С1-С6алкил), тогда R5c не представляет собой атом водорода или незамещенную C1-С6алкильную группу;
или его фармацевтически приемлемую соль или сольват.
Предпочтительными соединениями формулы (IV) являются соединения, где R5c представляет собой возможно замещенную С1-С6алкильную группу, причем предпочтительным заместителем является -Yc-R6c. Когда R5c замещен 5- или 6-членным гетероароматическим кольцом, содержащим от 1 до 4 гетероатомов, предпочтительно, чтобы число гетероатомов в кольце составляло не более чем 2.
Соединения формулы (IV) описаны в WO 01/44170.
В четвертом воплощении настоящего изобретения антагонист Р2Х7-рецептора представляет собой соединение формулы
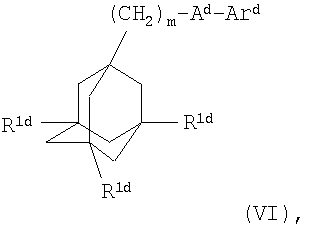
где m равно 1, 2 или 3;
каждый R1d независимо представляет собой атом водорода или галогена;
Аd представляет собой C(O)NH или NHC(O);
Ard представляет собой группу
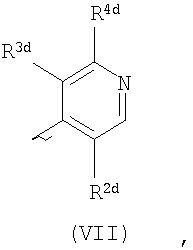 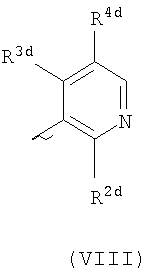 или или 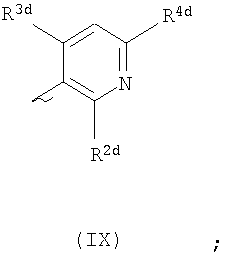
один из R2d и R3d представляет собой галоген, нитро, амино, гидроксил или группу, выбранную из (1) C1-С6алкила, возможно замещенного по меньшей мере одним атомом галогена, (2) С3-С8циклоалкила, (3) C1-С6алкокси, возможно замещенного по меньшей мере одним атомом галогена, и (4) С3-С8циклоалкилокси, а другой из R2d и R3d представляет собой атом водорода или галогена;
R4d представляет собой группу
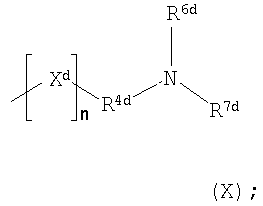
Хd представляет собой атом кислорода или серы, или группу >NR8d;
n равно 0 или 1;
R5d представляет собой С1-С5алкильную группу, которая может быть возможно замещена по меньшей мере одним заместителем, выбранным из гидроксила, галогена и С1-С6алкокси;
каждый из R6d и R7d независимо представляет собой атом водорода, C1-С6алкил (возможно замещенный по меньшей мере одним заместителем, выбранным из гидроксила, галогена и C1-С6алкокси и (ди)-С1-С4алкиламино (самого возможно замещенного по меньшей мере одной гидроксильной группой)) или С3-С8циклоалкил (возможно замещенный по меньшей мере одним заместителем, выбранным из гидроксила, галогена, С1-С6алкокси); и
R8d представляет собой атом водорода или С1-С5алкильную группу, которая может быть возможно замещена по меньшей мере одним заместителем, выбранным из гидроксила, галогена и С1-С6алкокси,
при условии, что:
(а) когда n равно 0, тогда Аd представляет собой NHC(O), и
(б) когда n равно 1, Xd представляет собой кислород, и Аd представляет собой С(O)NH, тогда R6d и R7d не представляют собой оба одновременно атом водорода или не представляют собой оба одновременно незамещенный C1-С6алкил, или когда один из R6d и R7d представляет собой атом водорода, тогда другой из R6d и R7d не представляет собой незамещенный С1-С6алкил, и
(в) когда n равно 1, Xd представляет собой кислород, серу или >NH, и Аd представляет собой NHC(O), тогда R6d и R7d не представляют собой оба одновременно атом водорода или не представляют собой оба одновременно незамещенный С1-С6алкил, или когда один из R6d и R7d представляет собой атом водорода, тогда другой из R6d и R7d не представляет собой незамещенный C1-С6алкил или -CH2CH2OH;
или его фармацевтически приемлемую соль или сольват.
Соединения формулы (VI) описаны в WO 03/41707.
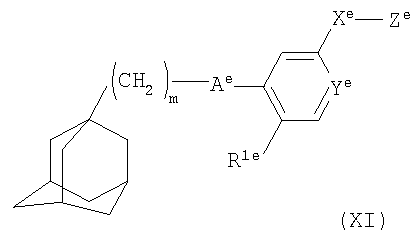
В другом аспекте настоящего изобретения антагонист Р2Х7-рецептора представляет собой соединение формулы
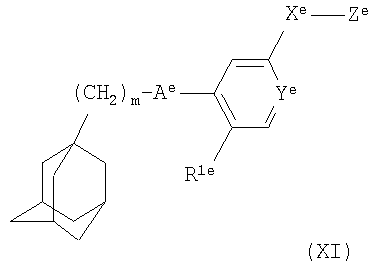
где m равно 1, 2 или 3;
Аe представляет собой С(O)NH или NHC(О);
Ye представляет собой N или СН;
Xе представляет собой связь, СО, (CH2)1-6, O(CH2)1-6, (CH2)1-6NH(CH2)1-6, (CH2)1-6O(CH2)1-6, NH(CH2)1-6;
Ze представляет собой NR2eR3e;
R1e представляет собой галоген, циано, нитро, амино, гидроксил, С1-С6алкил или С3-С8циклоалкил, причем алкильная или циклоалкильная группа может быть возможно замещена одним или более атомами фтора;
каждый из R2e и R3e независимо представляет собой атом водорода, C1-С6алкил или С3-С8циклоалкил, причем алкильная или циклоалкильная группа может быть возможно замещена одной или более группами, выбранными из гидроксила, галогена или C1-С6алкокси;
или R2e и R3e вместе с атомом азота, к которому они присоединены, образуют 3-9-членное насыщенное моно- или бициклическое гетероциклическое кольцо, содержащее от 1 до 2 атомов азота и возможно атом кислорода, которое может быть возможно замещено одной или более группами, выбранными из гидроксила, галогена или C1-С6алкокси;
или его фармацевтически приемлемую соль или сольват.
Соединения формулы (XI) могут быть получены химическими методами, соответствующими или аналогичными методам, описанным в ссылках, указанных выше.
В еще одном аспекте настоящего изобретения антагонист Р2Х7-рецептора представляет собой:
2-хлор-5-[[2-(2-гидрокси-этиламино)-этиламино]-метил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-[3-[(3-гидроксипропил)амино]пропил]-N-(трицикло[3.3.1.1]дец-1-илметил)бензамид,
(R)-2-хлор-5-[3-[(2-гидрокси-1-метилэтил)амино]пропил]-N-(трицикло[3.331.13,7]дец-1-илметил)-бензамид,
2-хлор-5-[[2-[(2-гидроксиэтил)амино]этокси]метил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-[3-[3-(метиламино)пропокси]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)бензамид,
2-хлор-5-[3-(3-гидроксипропиламино)-пропокси]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-[2-(3-гидроксипропиламино)этиламино]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-[2-(3-гидроксипропилсульфонил)этокси]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-[2-[2-[(2-гидроксиэтил)амино]этокси]этокси]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-[[2-[[2-(1-метил-1Н-имидазол-4-ил)этил]амино]этил]амино]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-пиперазин-1-илметил-N-(трицикло[3.3.1.1]дец-1-илметил)-бензамид,
2-хлор-5-(4-пиперидинилокси)-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-(2,5-диазабицикло[2.2.1]гепт-2-илметил)-N-(трицикло[3.3.1.1]дец-1-илметил)-бензамид,
2-хлор-5-(пиперидин-4-илсульфинил)-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
5-хлор-2-[3-[(3-гидроксипропил)амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамид,
2-хлор-5-[3-[[(1R)-2-гидрокси-1-метилэтил]амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-3-пиридинкарбоксамид,
5-хлор-2-[3-(этиламино)пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамид,
5-хлор-2-[3-[(2-гидроксиэтил)амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамид,
5-хлор-2-[3-[[(2S)-2-гидроксипропил)амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамид,
N-[2-метил-5-(9-окса-3,7-диазабицикло[3.3.1]нон-3-илкарбонил)фенил]трицикло[3.3.1.13,7]декан-1-ацетамид,
или фармацевтически приемлемую соль или сольват любого из них.
Фармацевтически приемлемые соли включают в себя, где целесообразно, соли присоединения кислоты, полученные с фармацевтически приемлемыми неорганическими и органическими кислотами, такие как хлорид, бромид, сульфат, фосфат, малеат, фумарат, тартрат, цитрат, бензоат, 4-метоксибензоат, 2- или 4-гидроксибензоат, 4-хлорбензоат, n-толуолсульфонат, метансульфонат, аскорбат, ацетат, сукцинат, лактат, глутарат, глюконат, трикарбаллилат, гидроксинафталин-карбоксилат или олеат; и соли, полученные с фармацевтически приемлемыми неорганическими или органическими основаниями. Соли, полученные с неорганическими основаниями, включают в себя соли алюминия, аммония, кальция, меди, железа трехвалентного, железа двухвалентного, лития, магния, марганца трехвалентного, марганца двухвалентного, калия, натрия, цинка и висмута. Особенно предпочтительными являются соли аммония, кальция, магния, калия и натрия. Соли, полученные с фармацевтически приемлемыми органическими основаниями, включают в себя соли первичных, вторичных и третичных аминов, циклических аминов, подобных аргинину, бетаину, холину и тому подобных. Примеры фармацевтически приемлемых сольватов включают в себя гидраты.
Примеры антагонистов Р2Х7-рецептора, которые могут быть использованы в настоящем изобретении, включают в себя:
2-хлор-5-[[2-(2-гидрокси-этиламино)-этиламино]-метил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид дигидрохлорид,
2-хлор-5-[3-[(3-гидроксипропил)амино]пропил]-N-(трицикло[3.3.1.1]дец-1-илметил)бензамид гидрохлорид,
(R)-2-хлор-5-[3-[(2-гидрокси-1-метилэтил)амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-млметил)-бензамид гидрохлорид,
2-хлор-5-[[2-[(2-гидроксиэтил)амино]этокси]метил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид ацетат (1:1),
2-хлор-5-[3-[3-(метиламино)пропокси]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)бензамид гидрохлорид,
2-хлор-5-[3-(3-гидрокси-пропиламино)-пропокси]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид гидрохлорид,
2-хлор-5-[2-(3-гидроксипропиламино)этиламино]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид ацетат (1:1),
2-хлор-5-[2-(3-гидроксипропилсульфонил)этокси]-N-(трицикло[3.3.1.13,7]дец-1-илметил)бензамид,
2-хлор-5-[2-[2-[(2-гидроксиэтил)амино]этокси]этокси]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид гидрохлорид,
2-хлор-5-[[2-[[2-(1-метил-1H-имидазол-4-ил)этил]амино]этил]амино]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-пиперазин-1-илметил-N-(трицикло[3.3.1.1]дец-1-илметил)бензамид дигидрохлорид,
2-хлор-5-(4-пиперидинилокси)-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид гидрохлорид,
2-хлор-5-(2,5-диазабицикло[2.2.1]гепт-2-илметил)-N-(трицикло[3.3.1.1]дец-1-илметил)-бензамид гидрохлорид,
2-хлор-5-(пиперидин-4-илсульфинил)-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
5-хлор-2-[3-[(3-гидроксипропил)амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамид,
2-хлор-5-[3-[[(1R)-2-гидрокси-1-метилэтил]амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-3-пиридинкарбоксамид,
5-хлор-2-[3-(этиламино)пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамид гидрохлорид,
5-хлор-2-[3-[(2-гидроксиэтил)амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамид гидрохлорид,
5-хлор-2-[3-[[(25)-2-гидроксипропил)амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамиддигидрохлорид и
N-[2-метил-5-(9-окса-3,7-диазабицикло[3.3.1]нон-3-илкарбонил)фенил]трицикло[3.3.1.13,7]декан-1-ацетамид гидрохлорид.
Активные ингредиенты, используемые в настоящем изобретении, могут быть способны существовать в стереоизомерных формах. Следует понимать, что изобретение охватывает все геометрические и оптические изомеры активных ингредиентов и их смеси, включая рацематы. Таутомеры и их смеси также образуют аспект настоящего изобретения.
Второй активный ингредиент согласно настоящему изобретению представляет собой нестероидное противовоспалительное лекарственное средство (НСПВС). НСПВС представляет собой соединение или вещество, которые способны ингибировать, полностью либо частично, фермент циклооксигеназу (СОХ). Этот фермент имеет по меньшей мере две изоформы, обозначаемые как СОХ-1, которая экспрессируется конститутивно и служит для защиты выстилающих тканей желудка и кишечника, и СОХ-2, которая является индуцибельной и играет важную роль в воспалительном процессе. Селективные ингибиторы СОХ-2 также известны как COXIB.
НСПВС по изобретению может ингибировать как СОХ-1, так и СОХ-2, но предпочтительно селективен в отношении СОХ-2.
Примеры НСПВС, которые могут быть использованы, включают в себя ибупрофен, напроксен, аспирин, целекоксиб (коммерчески доступен под товарным знаком "Celebrex"), диклофенак (коммерчески доступен под товарным знаком "Voltaren"), этодолак (коммерчески доступен под товарным знаком "Lodine"), фенопрофен (коммерчески доступен под товарным знаком "Nalfon"), индометацин (коммерчески доступен под товарным знаком "Indocin"), кетопрофен (коммерчески доступен под товарным знаком "Oruvail"), кеторалак (коммерчески доступен под товарным знаком "Toradol"), оксапрозин (коммерчески доступен под товарным знаком "Daypro"), набуметон (коммерчески доступен под товарным знаком "Relafen"), сулиндак (коммерчески доступен под товарным знаком "Clinoril"), толметин (коммерчески доступен под товарным знаком "Tolectin"), рофекоксиб (коммерчески доступен под товарным знаком "Vioxx"), вальдекоксиб, лумарикоксиб, мелоксикам, эторикоксиб и парекоксиб.
В воплощении изобретения второй активный ингредиент представляет собой селективный ингибитор СОХ-2. В контексте этого воплощения селективный ингибитор СОХ-2 представляет собой соединение, которое демонстрирует селективность in vitro для СОХ-2: СОХ-1, составляющую по меньшей мере 2:1, при измерении в анализе цельной крови, как описано Warner, Т.О. et at., Proc. Natl. Acad. Sci. USA, 1999, 96, 7563-7568. Предпочтительно селективный ингибитор СОХ-2 имеет селективность in vitro для СОХ-2: СОХ-1 по меньшей мере 5:1, более предпочтительно по меньшей мере 10:1, еще более предпочтительно по меньшей мере 30:1 и наиболее предпочтительно по меньшей мере 100:1. Примеры селективных ингибиторов СОХ-2, которые могут быть использованы в соответствии с этим. воплощением, включают в себя целекоксиб, рофекоксиб, вальдекоксиб, лумарикоксиб, эторикоксиб и парекоксиб.
В одном воплощении настоящего изобретения второй активный ингредиент представляет собой селективный ингибитор СОХ-2 целекоксиб. Химическое название целекоксиба 4-[5-(4-метилфенил)-3-(трифторметил)-1H-пиразол-1-ил]бензолсульфонамид (Penning, Т. et al., J. Med. Chem., 1997, 40, 1347-1365). Целекоксиб производится Pfizer под товарным знаком "Celebrex".
В одном воплощении настоящего изобретения второй активный ингредиент представляет собой селективный ингибитор СОХ-2 рофекоксиб. Химическое название рофекоксиба 4-[4-(метилсульфонил)фенил]-3-фенил-(5Н)-фуранон (Chan, C.C. et al., J. Pharmacol. Exp., Ther., 1999, 290, 551-560). Рофекоксиб производится Merck Sharp&Dohme под товарным знаком "Vioxx".
В другом воплощении настоящего изобретения второй активный ингредиент представляет собой селективный ингибитор СОХ-2 вальдекоксиб. Химическое название вальдекоксиба 4-(5-метил-3-фенил-4-изоксазолил)-бензолсульфонамид (Talley, J.J. et al., J. Med. Chem., 2000, 43, 775-777). Вальдекоксиб производится Pfizer под товарным знаком "Bextra".
Было установлено, что выбранная группа активных ингредиентов согласно изобретению является полезной, поскольку имеет результатом целебный противовоспалительный эффект, и, следовательно, может быть использована для лечения различных острых и хронических воспалительных состояний/расстройств, таких как ревматоидный артрит и остеоартрит. Лечение воспалительных расстройств может вовлекать уменьшение опухоли и/или облегчение боли, связанной с этим состоянием. В этом отношении продукты по настоящему изобретению проявили себя как особенно эффективные в ослаблении или облегчении боли, вызванной воспалительными расстройствами суставов.
Фармацевтическая композиция по изобретению может быть приготовлена смешиванием первого активного ингредиента со вторым активным ингредиентом. Соответственно, в дополнительном аспекте настоящего изобретения предлагается способ получения фармацевтической композиции, включающий смешивание первого активного ингредиента, который представляет собой антагонист Р2Х7-рецептора, со вторым активным ингредиентом, который представляет собой нестероидное противовоспалительное лекарственное средство.
Альтернативно, для лечения воспалительных состояний первый и второй активные ингредиенты могут быть введены одновременно (иначе чем в виде смеси, описанной выше), последовательно или раздельно. "Последовательно" означает, что первый и второй активные ингредиенты вводят в любом порядке, непосредственно один за другим. Они еще будут оказывать требуемый эффект, если их вводят раздельно, но с интервалом менее 4 часов, предпочтительно с интервалом менее 2 часов, более предпочтительно с интервалом менее 30 минут.
Соответственно, согласно изобретению также предложен фармацевтический продукт, содержащий в комбинации препарат первого активного ингредиента, который представляет собой антагонист Р2Х7-рецептора, и препарат второго активного ингредиента, который представляет собой нестероидное противовоспалительное лекарственное средство, для одновременного, последовательного или раздельного использования в терапии. Второй активный ингредиент предпочтительно представляет собой селективный ингибитор СОХ-2.
В другом аспекте изобретения предложен набор, содержащий препарат первого активного ингредиента, который представляет собой антагонист Р2Х7-рецептора, препарат второго активного ингредиента, который представляет собой нестероидное противовоспалительное лекарственное средство, и инструкции по одновременному, последовательному или раздельному введению этих препаратов нуждающемуся в этом пациенту.
Первый и второй активные ингредиенты традиционно вводят посредством перорального или парентерального введения, используя традиционные системные лекарственные формы, такие как таблетки, капсулы, пилюли, порошки, водные или масляные растворы или суспензии, эмульсии и стерильные водные или масляные растворы или суспензии для инъекций. Эти лекарственные формы, как правило, включают в себя один или более фармацевтически приемлемых ингредиентов, которые могут быть выбраны, например, из адъювантов, носителей, связующих веществ, скользящих веществ, разбавителей, стабилизаторов, забуферивающих агентов, эмульгаторов, регуляторов вязкости, поверхностно-активных веществ, консервантов, корригентов и красителей. Предпочтительно первый и второй активные ингредиенты доставляют перорально.
Для вышеуказанных терапевтических применений вводимые дозировки, конечно, будут отличаться в зависимости от используемых первого и второго активных ингредиентов, способа введения, необходимого лечения и установленного состояния или расстройства. Однако, в целом, удовлетворительные результаты получают, когда общая, комбинированная, суточная дозировка первого и второго активных ингредиентов при пероральном введении лежит в диапазоне от 10 до 2000 миллиграмм (мг), предпочтительно от 10, 20, 30, 40, 50, 100, 150, 200 или 300 до 1800, 1500, 1200, 1000, 800, 700, 600, 500 или 400 мг.
Фармацевтическая композиция, фармацевтический продукт или набор по изобретению могут быть введены в виде разделенных доз от 1 до 4 раз в сутки и, предпочтительно, один или два раза в сутки.
В воплощении настоящего изобретения суточная дозировка первого активного ингредиента в фармацевтической композиции, продукте или наборе лежит в диапазоне от 5 до 1000 мг, от 5 до 800 мг, от 5 до 600 мг, от 5 до 500 мг, от 5 до 400 мг, от 5 до 300 мг, от 5 до 200 мг, от 5 до 100 мг, от 5 до 50 мг, от 20 до 1000 мг, от 20 до 800 мг, от 20 до 600 мг, от 20 до 500 мг, от 20 до 400 мг, от 20 до 300 мг, от 20 до 200 мг, от 20 до 100 мг, от 20 до 50 мг, от 50 до 1000 мг, от 50 до 800 мг, от 50 до 600 мг, от 50 до 500 мг, от 50 до 400 мг, от 50 до 300 мг, от 50 до 200 мг, от 50 до 100 мг, от 100 до 1000 мг, от 100 до 800 мг, от 100 до 600 мг, от 100 до 500 мг, от 100 до 400 мг, от 100 до 300 мг, от 100 до 200 мг, тогда как суточная доза второго активного ингредиента лежит в диапазоне от 1 до 200 мг, от 1 до 100 мг, от 1 до 50 мг, от 1 до 25 мг, от 5 до 200 мг, от 5 до 100 мг, от 5 до 50 мг, от 5 до 25 мг, от 10 до 200 мг, от 10 до 100 мг, от 10 до 50 мг или от 10 до 25 мг; причем суточные дозы первого и второго активных ингредиентов могут быть введены в виде разделенных доз от 1 до 4 раз в сутки, предпочтительно один или два раза в сутки, причем первый и второй активные ингредиенты могут быть введены в смеси, одновременно, последовательно или раздельно. Схема приема согласно этому воплощению с легкостью может быть подобрана для случая, когда как первый, так и второй активные ингредиенты доставляют путем перорального введения. Вторые активные ингредиенты, которые могут быть использованы согласно этому воплощению, включают целекоксиб, рофекоксиб и вальдекоксиб.
Кроме того, согласно настоящему изобретению предложено применение фармацевической композиции по изобретению для изготовления лекарства для лечения воспалительного расстройства, в частности ревматоидного артрита или остеоартрита.
Согласно настоящему изобретению также предложен способ лечения воспалительного расстройства, который включает введение терапевтически эффективного количества фармацевтической композиции по изобретению нуждающемуся в этом пациенту, при этом конкретными воспалительными расстройствами являются ревматоидный артрит или остеоартрит.
Кроме того, согласно настоящему изобретению также предложен способ лечения воспалительного расстройства, который включает одновременное, последовательное или раздельное введение:
(а) (терапевтически эффективной) дозы первого активного ингредиента, который представляет собой антагонист Р2Х7-рецептора, и
(б) (терапевтически эффективной) дозы второго активного ингредиента, который представляет собой нестероидное противовоспалительное лекарственное средство, нуждающемуся в этом пациенту.
В контексте настоящего изобретения термин "терапия" также включает в себя "профилактику", если не оговорено противное. Термины "терапевтический" и "терапевтически" следует истолковывать соответственно.
Профилактика, как следует ожидать, является особенно уместной для лечения лиц, которые имели предшествующий случай рассматриваемого состояния или расстройства или же, как полагают, имеют повышенный риск рассматриваемого состояния или расстройства. Лица, имеющие риск развития конкретного состояния или расстройства, обычно включают лиц, имеющих семейный анамнез этого состояния или расстройства, или лиц, которые, как было установлено с помощью генетического тестирования или скрининга, являются особенно восприимчивыми к развитию данного состояния или расстройства.
Кроме того, изобретение относится к терапиям на основе тройных комбинаций для лечения любого из перечисленных расстройств: ревматоидный артрит, остеоартрит, остеопороз, псориаз, воспалительные заболевания кишечника, ХОЗЛ (хроническое обструктивное заболевание легких), астма, аллергический ринит или рак, или нейродегенеративные болезни, такие как рассеянный склероз, болезнь Альцгеймера или инсульт.
Для лечения ревматоидного артрита фармацевтическая композиция по изобретению может быть скомбинирована с такими "биологическими агентами" как антагонисты рецептора IL-1 (например анакинра) и IL-1 trap, рецептора IL-18, анти-IL-6 Ab, анти-СО-20 Ab, анти-IL-15 Ab и CTLA4Ig.
Подходящие агенты, которые могут быть использованы в комбинации с фармацевтической композицией по изобретению, включают в себя доноры оксида азота, ингибирующие циклооксигеназу (CINOD), и "модифицирующие заболевание агенты" (DMARD), такие как циклоспорин А, лефлюномид; циклесонид; гидроксихлороквин, d-пеницилламин, ауранофин или парентеральное или пероральное золото также могут быть использованы.
Кроме того, настоящее изобретение также относится к комбинации фармацевтической композиции по изобретению с ингибитором биосинтеза лейкотриена, ингибитором 5-липоксигеназы (5-LO) или антагонистом белка, активирующего 5-липоксигеназу (FLAP), выбранным из группы, образованной зилеутоном; АТ-761; фенлеутоном; тепоксалином; Abhott-79175; Abbott-85761; N-(5-замещенными)тиофен-2-алкилсульфонамидами; 2,6-ди-третбутил-фенолгидразонами; метокситетрагидропиранами, такими как Zeneca ZD-2138, соединением SB-210661; пиридинил-замещенными 2n цианонафталиновыми соединениями, такими как L-739,010; 2-цианохинолиновыми соединениями, такими как L-746,530; индольными и хинолиновыми соединениями, такими как МК-591, МК-886 и BAY x 1005.
Кроме того, настоящее изобретение также относится к фармацевтической композиции по изобретению совместно с антагонистом рецептора лейкотриенов LTB4, LTC4, LTD4 и LTE4, выбранным из группы, состоящей из фенотиазин-3-онов, таких как L-651,392; амидиновых соединений, таких как CGS-25019C; бензоксаламинов, таких как онтазоласт; бензолкарбоксимидамидов, таких как BIIL284/260; и таких соединений как зафирлукаст, аблукаст, монтелукаст, пранлукаст, верлукаст (МК-679), RG-12525, Ro-245913, иралукаст (CGP 45715А) и BAY x 7195.
Кроме того, настоящее изобретение также относится к фармацевтической композиции по изобретению совместно с ингибитором фосфодиэстеразы-4 (PDE4), включая ингибиторы изоформы PDE4D.
Кроме того, настоящее изобретение также относится к фармацевтической композиции по изобретению совместно с антигистаминными антагонистами Н1-грецепторов, включая цетиризин, лоратадин, дезлоратадин, фексофенадин, астемизол, азеластин и хлорфенирамин.
Кроме того, настоящее изобретение также относится к фармацевтической композиции по изобретению совместно с гастропротективным антагонистом Н2-рецепторов или ингибиторами протонного насоса (такими как омепразол).
Кроме того, настоящее изобретение также относится к фармацевтической композиции по изобретению совместно с сосудосуживающим симпатомиметиком, являющимся агонистом 1- и 2-адренорецепторов, включая пропилгекседрин, фенилэфрин, фенилпропаноламин, псевдоэфедрин, нафазолина гидрохлорид, оксиметазолина гидрохлорид, тетрагидрозолина гидрохлорид, ксилометазолина гидрохлорид и этилнорепинфрина гидрохлорид.
Кроме того, настоящее изобретение также относится к фармацевтической композиции по изобретению совместно с антихолинэргическими агентами, включая ипратропиум бромид; тиотропиум бромид; окситропиум бромид; пирензепин и телензепин.
Кроме того, настоящее изобретение также относится к фармацевтической композиции по изобретению совместно с метилксантанинами, включая теофиллин и аминофиллин; натрия хромогликат или антагонист мускариновых рецепторов (М1, М2 и М3).
Кроме того, настоящее изобретение также относится к фармацевтической композиции по изобретению совместно с модуляторами функции хемокиновых рецепторов, таких как CCR1, CCR2, CCR2A, CCR2B, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9, CCR10 и CCR11 (для С-С семейства), CXCR1, CXCR3, CXCR4 и CXCR5 (для С-Х-С семейства) и СХ3СR1 для С-Х3-С семейства.
Кроме того, настоящее изобретение также относится к фармацевтической композиции по изобретению совместно с миметиком инсулинподобного фактора роста типа I (IGF-1).
Кроме того, настоящее изобретение также относится к фармацевтической композиции по изобретению совместно с (а) ингибиторами триптазы; (б) антагонистами фактора активации тромбоцитов (PAF); (в) ингибиторами интерлейкин-конвертирующего фермента (ICE); (г) ингибиторами IMPDH (инозинмонофосфатдегидрогеназа); (д) ингибиторами молекул адгезии, включая антагонисты VLA-4; (е) катепсинами; (ж) ингибиторами глюкозо-6-фосфатдегидрогеназы; (з) антагонистами кинин-B1- и В2-рецепторов; (и) противоподагрических агентов, например колхицина; (к) ингибиторами ксантиноксидазы, например аллопуринолом; (л) урикозурическими агентами, например пробенецидом, сульфинпиразоном и бензбромароном; (м) стимуляторами секреции гормона роста; (н) трансформирующим фактором роста (TGF); тромбоцитарным фактором роста (PDGF); фактором роста фибробластов, например основным фактором роста фибробластов (bFTF); (о) гранулоцитарно-макрофагальным колониестимулирующим фактором (GM-CSF); (п) капсаициновым кремом; (р) антагонистами тахикининовых NK1- и NK2-рецепторов, выбранными из группы, состоящей из NKP-608C; SB-233412 (талнетант) и D-4418; и (с) ингибиторами эластазы, выбранными из группы, состоящей из UT-77 и ZD-0892; (т) ингибиторами индуцибельной синтазы оксида азота (iNOS) или (у) молекулой, гомологичной рецептору хемоаттрактанта, экспрессирующемуся на ТН2-клетках (антагонистами CRTH2).
Фармацевтическая композиция по изобретению также может быть использована в комбинации с существующими терапевтическими агентами для лечения остеоартрита. Подходящие агенты, которые могут быть использованы в комбинации, включают в себя ингибиторы индуцибельной синтазы оксида азота (iNOS ингибиторы) и доноры оксида азота, - ингибирующие циклооксигеназу (CINOD), аналгетики (такие как парацетамол и трамадол), хондропротекторные агенты, такие как диацереин, доксицилин и глюкозамин и гиалуроновые кислоты, такие как гиалган и синвиск.
Фармацевтическая композиция по изобретению также может быть использована в комбинации с существующими терапевтическими агентами для лечения воспалительных заболеваний кишечника (неспецифический язвенный колит и болезнь Крона). Подходящие агенты, которые могут быть использованы, включают в себя 5-аминосалицилаты, тиопурины, азатиоприн и 6-меркаптопурин.
Фармацевтическая композиция по изобретению также может быть использована в комбинации с противораковыми агентами, такими как эндостатин и ангиостатин, или цитотоксическими лекарствами, такими как адриамицин, дауномицин, цис-платина, этопозид, таксол, таксотер и ингибиторы фарнезилтрансферазы, ингибиторы VegF и антиметаболиты, такие как антинеопластические агенты, особенно антимитотические лекарства, включающие в себя алкалоиды барвинка розового, такие как винбластин и винкристин.
Фармацевтическая композиция по изобретению также может быть использована в комбинации с противовирусными агентами, такими как вирацепт, AZT, ацикловир и фамцикловир, и антисептическими соединениями, такими как валант.
Фармацевтическая композиция по изобретению также может быть использована в комбинации с блокаторами кальциевых каналов, агентами, снижающими липиды, такими как фибраты, бета-блокаторы, ингибиторы АПФ (ангиотензин-превращающий фермент), антагонистами ангиотензиновых рецепторов 2-го типа и ингибиторами агрегации тромбоцитов.
Фармацевтическая композиция по изобретению также может быть использована в комбинации с агентами с активностью в отношении ЦНС, такими как антидипрессанты (такими как сертралин), антипаркинсонические лекарства (такие как депренил, леводопа (L-допа), реквип, мирапекс, ингибиторы моноаминооксидазы В (МАО-В), такие как селегин и расагилин, ингибиторы субстанции Р (comP), такие кактасмар, ингибиторы А-2, ингибиторы обратного захвата дофамина, антагонисты NMDA (N-метил-D-аспарагиновая кислота), агонисты никотина, агонисты дофамина и ингибиторы нейрональной NO-синтазы) и лекарства против болезни Альцгеймера, такие как донепезил, такрин, пропентофиллин или метрифонат.
Фармацевтическая композиция по изобретению также может быть использована в комбинации с противоостеопорозными агентами, такими как ролоксифен, дролоксифен, лазофоксифен или фосомакс, и иммунодепрессантами, такими как FK-506, рапамицин, циклоспорин и азатиоприн.
Настоящее изобретение далее будет пояснено посредством ссылок на нижеследующие иллюстрирующие примеры.
В данных примерах были использованы следующие Р2Х7-антагонисты.
1. N-[2-метил-5-(9-окса-3,7-диазабицикло[3.3.1.1]нон-3-илкарбонил)-фенил]трицикло[3.3.1.13,7]декан-1-ацетамид гидрохлорид
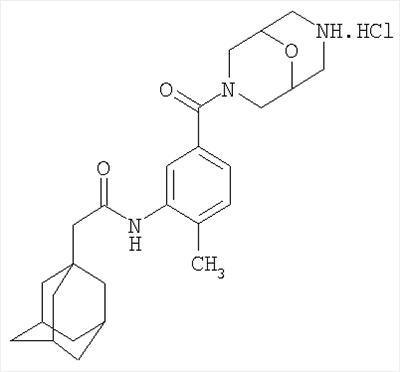
Р2Х7-антагонист 1. (N-[2-метил-5-(9-окса-3,7-диазабицикло[3.3.1.1]нон-3-млкарбонил)фенил]трицикло[3.3.1.13,7]декан-1-ацетамид гидрохлорид) получали следующим образом:
а) 3-(4-метил-3-нитробензоил)-7-(фенилметил)-9-окса-3,7-диазаби-цикло[3.3.1.1]нонан
Оксалилхлорид (9,6 мл) в дихлорметане (30 мл) добавляли по каплям в течение 45 минут к охлажденному на льду раствору 4-метил-3-нитро-бензойной кислоты (10,0 г) в дихлорметане (320 мл), содержащему ДМФ (N,N-диметилформамид) (0,1 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, а затем концентрировали под вакуумом. Хлорангидрид переносили в ТГФ (тетрагидрофуран) (320 мл) и охлаждали на ледяной бане перед добавлением N,N-диизопропилэтиламина (38 мл), а затем порций дигидрохлорида 3-(фенилметил)-9-окса-3,7-диаза-бицикло[3.3.1.1]нонана (16,0 г) (полученного как описано в WO 01/028992). Реакционную смесь перемешивали в течение 18 часов, затем ее разбавляли этилацетатом (600 мл) и промывали водой (2×200 мл) и насыщенным раствором бикарбоната натрия (водный) (3×150 мл), затем сушили (MgSO4), фильтровали и концентрировали с получением указанного в подзаголовке соединения (18,5 г).
m/z=382
б) 3-(3-амино-4-метилбензоил)-7-(фенилметил)-9-окса-3,7-диазабицикло[3.3.1.1]нонан
Порошок восстановленного железа (7,9 г) добавляли в течение 15 минут к перемешиваемому раствору продукта, полученного на этапе (а) (18,0 г), и хлорида аммония (7,5 г) в смеси этанол/вода (3:1, 320 мл) при 70°С. Реакционную смесь нагревали с обратным холодильником в течение 2 часов, затем фильтровали и концентрировали под вакуумом. Остаток переносили в этилацетат (400 мл), промывали водой (2×150 мл), затем органическую фазу сушили (MgSO4) и концентрировали под вакуумом с получением указанного в подзаголовке соединения (14,5 г).
m/z=352
в) N-[2-метил-5-[[7-(фенилметил)-9-окса-3,7-диазабицикло[3.3.1.1]нон-3-ил]карбонил]фенил]трицикло[3.3.1.13,7]декан-1-ацетамид гидрохлорид
Получали способом этапа (а), используя 1-адамантануксусную кислоту и продукт этапа (б). Перекристаллизация (этилацетат) позволила получить указанное в подзаголовке соединение.
m/z=528
г) N-[2-метил-5-(9-окса-3,7-диазабицикло[3.3.1.1]нон-3-илкарбонил)фенил]трицикло[3.3.1.13,7]декан-1-ацетамид гидрохлорид
4 М HCl в 1,4-диоксане (8 мл) добавляли к раствору продукта этапа (в) (13,0 г) в этилацетате (300 мл). Полученный в результате осадок отделяли фильтрованием, затем суспендировали в этаноле (300 мл) и добавляли 5% палладий на углероде (1,2 г). Реакционную смесь перемешивали под давлением водорода 3 атмосферы (примерно 300 кПа) в течение 36 часов. Затем в атмосфере азота добавляли метанол, затем катализатор удаляли фильтрованием, фильтрат концентрировали под вакуумом и перекристаллизовывали (смесь изопропанол:метанол, 25:1, 800 мл) с получением указанного в заголовке соединения (9,1 г).
m/z=438 (M+H)+
 H (400 МГц, d6-ДМСО, Me4Si, 90°С) 9.06 (1Н, s), 7.64 (1H, s), 7.25 (1H, m), 7.19 (1H, m), 4.15 (2H, s), 3.96 (2H, d, J=14 Гц), 3.35-3.23 (6Н, m), 2.26 (3H, s), 2.14 (2H,s), 1.96 (3H, brs), 1.69-1.62 (12H, m). H (400 МГц, d6-ДМСО, Me4Si, 90°С) 9.06 (1Н, s), 7.64 (1H, s), 7.25 (1H, m), 7.19 (1H, m), 4.15 (2H, s), 3.96 (2H, d, J=14 Гц), 3.35-3.23 (6Н, m), 2.26 (3H, s), 2.14 (2H,s), 1.96 (3H, brs), 1.69-1.62 (12H, m).
Пример 1
Фармакологический анализ для определения эффекта комбинаций НСПВС/Р2Х7-антагонист (без добавления Р2Х7-агониста)
Моноциты периферической крови человека были получены из крови здоровых добровольцев, которую собирали в пробирки для крови с ЭДТА (этилендиаминтетрауксусная кислота). Моноциты выделяли с помощью последовательного градиентного центрифугирования и промывания с получением чистой популяции клеток. Затем к клеточной суспензии в тканевой культуре добавляли липополисахарид (ЛПС) и инкубировали в течение 4-12 часов при 37°С. Затем к клеткам добавляли НСПВС и/или Р2Х7-антагонист или носитель. После инкубации образцы клеточных супернатантов переносили в 96-луночный планшет для последующего определения цитокинов и медиаторов. Образование воспалительных медиаторов измеряли в клеточных супернатантах с помощью специфичных ELISA анализов для цитокинов IL-1, IL-18, TNF- и для других медиаторов включая простагландин Е2 (PGE2), N0 и матриксные металлопротеиназы (ММР). Определяли уровни медиаторов, высвобожденных в присутствии только антагониста Р2Х7-рецептора, или в присутствии только НСПВС или в присутствии комбинации антагониста Р2Х7-рецептора с НСПВС. Затем сравнивали эффекты антагонистов и НСПВС по отдельности и в комбинации. Статистически значимые уровни ингибирующей активности комбинаций Р2Х7-антагонист/НСПВС в отношении одного медиатора (IL-1 или TNF-) или нескольких медиаторов по сравнению с уровнями, достигнутыми либо только Р2Х7-антагонистом, либо только НСПВС, являются показателем повышенной эффективности при лечении заболевания.
Пример 2
Фармакологический анализ для определения эффекта комбинаций НСПВС/Р2Х7-антагонист (с добавлением Р2Х7-агониста)
Моноциты периферической крови человека были получены из крови здоровых добровольцев, которую собирали в пробирки для крови с ЭДТА. Моноциты выделяли с помощью последовательного градиентного центрифугирования и промывания с получением чистой популяции клеток. Затем к клеточной суспензии в тканевой культуре добавляли липополисахарид (ЛПС) и инкубировали в течение 4-12 часов при 37°С. Затем добавляли тестируемые смеси с последующим добавлением агониста Р2Х7-рецептора BzATP (3-O-(4-бензоил)бензоил-аденозин-5-трифосфат). Тестируемые смеси могут содержать носитель в качестве контроля, антагонист Р2Х7-рецептора или комбинацию антагониста Р2Х7-рецептора с НСПВС. После инкубации образцы клеточных супернатантов переносили в 96-луночный планшет для последующего определения цитокинов и медиаторов. Образование воспалительных медиаторов измеряли в клеточных супернатантах с помощью специфичных ELISA анализов на цитокины IL-1, IL-18, TNF- и на другие медиаторы, включая PGE2, NO и матриксные металлопротеиназы (ММР). Определяли уровни медиаторов, высвобожденных в присутствии только антагониста Р2Х7-рецептора или в присутствии комбинации антагониста Р2Х7-рецептора с НСПВС. Затем сравнивали эффекты, продуцируемые Р2Х7-антагонистом по отдельности и в комбинации с НСПВС. Статистически значимые уровни ингибирующей активности комбинаций Р2Х7-антагонист/НСПВС в отношении одного медиатора (IL-1 или TNF-) или нескольких медиаторов по сравнению с уровнями, достигнутыми только Р2Х7-антагонистом, являются показателем повышенной эффективности при лечении заболевания.
Пример 3А
Оценка противовоспалительной активности комбинаций ингибитор СОХ-2, целекоксиб/Р2Х7-антагонист в модели артрита, индуцированного у крыс клеточной стенкой стрептококка1
Артрит индуцировали клеточной стенкой стрептококка (SCW) в левом голеностопном суставе самок крыс линии Lewis. Животных сенсибилизировали внутрисуставной инъекцией 5 мкг (в 5 мкл) SCW (Lee Laboratories) в левый голеностопный сустав. Опухание левого голеностопного сустава оценивали через 3 суток после инъекции и нереагирующих животных (животных с отсутствием видимого опухания голеностопного сустава) исключали. Реагирующие животные были случайным образом распределены по группам тестирования.
Артрит индуцировали на 21-е сутки после сенсибилизации с помощью внутривенной (в.в.) инъекции SCW (100 мкг в 500 мкл физиологического раствора). Животных контролировали и оценивали ежедневно до истечения 6 суток после индукции. Крыс содержали на опилках со свободным доступом к пище и воде.
В этом примере Р2Х7-антагонист 1 вводили перорально в дозе 30 мг/кг (4 мл/кг, два раза в сутки). Данное соединение вводили в виде суспензии в 1%-ной (мас./об.) метилцеллюлозе в деионизированной воде, свежие партии которой готовили ежедневно. Введение доз начинали за сутки до индукции артрита и продолжали ежедневно до истечения 6 суток после индукции. Целекоксиб (3 мг/кг) вводили перорально по такой же схеме, как и для Р2Х7-антагониста 1, причем введение целекоксиба проводили немедленно после введения Р2Х7-антагониста 1.
Диаметр голеностопного сустава измеряли с помощью штангенциркуля с нониусом ежедневно, начиная с суток -1. Механические пороги чувствительности оценивали, используя щетинки вон Фрея, на сутки -1, 1, 3 и 5. Воздействие щетинками наносили в возрастающей жесткости в область голеностопного сустава подушечек лап обоих ступней. Первую щетинку, которая индуцировала эффект отдергивания, считали порогом.
Эффекты в отношении опухоли голеностопного сустава и механический порог рассчитывали по площади под кривой (AUC) как сумму отклонений от индивидуальных значений на сутки -1. Рассчитывали величину и направленность взаимодействия и осуществляли анализ данных выполняли с помощью дисперсионного анализа (ANOVA), с последующим применением критерия Дунетта для AUC-данных (версия SAS 8.01). Результаты представлены в таблице 1.
| Таблица 1 |
| |
% снижения AUC (по сравнению с контролем-носителем для артрита) |
| Опухоль голеностопного сустава |
Порог вон Фрея |
| Р2Х7-антагонист 1 |
28,5±13,5 |
21,1±10,9 |
| Целекоксиб |
63,0±3,9** |
43,2±15,9* |
| Р2Х7-антагонист 1 + целекоксиб |
59,4±6,2** |
64,2±10,3** тестирование взаимодействия р=1,00*** |
| * р<0,01, ** р<0,001 по сравнению с контролем-носителем для артрита, |
| *** величина взаимодействия, указывающая на аддитивный эффект комбинации. |
Из вышеуказанных результатов можно видеть, что комбинация Р2Х7-антагониста 1 и целекоксиба показала позитивное взаимодействие, проявлявшееся в снижении механического порога.
В последующих исследованиях Р2Х7-антагонист 1 вводили в дозах 10 и 30 мг/кг в комбинации в целекоксибом в дозе 1, 3 и 10 мг/кг, где два активных ингредиента вводили совместно в едином препарате. Экспериментальные конечные точки были такими же, как описано ранее. Результаты этих исследований подтвердили позитивное взаимодействие, проявлявшееся в снижении механического порога как описано выше. Кроме того, анализ образцов крови из этих исследований продемонстрировал, что фармакокинетические профили двух лекарств при их введении в комбинации были идентичны профилям при их индивидуальном введении. Это указывает на то, что обнаруженные позитивные эффекты не могут быть приписаны изменениям в фармакокинетических профилях лекарств, а являются результатом фармакологического взаимодействия.
Обнаружение того, что Р2Х7-антагонист 1 и целекоксиб оказывают позитивный эффект в отношении порога вон Фрея в комбинации, которая демонстрирует незначительное благоприятное действие в отношении опухоли голеностопного сустава, указывает на то, что эта комбинация лекарств обладает сильным и неожиданно позитивным эффектом в отношении боли в воспаленном суставе.
1. Методика эксперимента основана на методике, описанной в Carlson R.P., Jacobsen P.В.; Comparison of adjuvant and streptococcal cell wall-induced arthritis in the rat in Morgan D.W., Marshall L.A., editors; In vivo models of Inflammation. Basel: Birkhauser Verlag; 1999).
Пример 3Б
Оценка противовоспалительной активности комбинаций ингибитор СОХ-2, рофекоксиб/Р2Х7-антагонист в модели артрита, индуцированного у крыс клеточной стенкой стрептококка1
Противовоспалительную активность ингибитора СОХ-2, рофекоксиба, в комбинации с Р2Х7-антагонистом оценивали, используя протокол, описанный в примере 3А. Р2Х7-антагонист 1 вводили перорально в дозе 30 мг/кг (4 мл/кг, два раза в сутки) в виде суспензии в 1%-ной (мас./об.) метилцеллюлозе в деионизированной воде вместе с рофекоксибом (Merck Sharp & Dohme Limited) (1 мг/кг) в виде единого препарата. Введение доз начинали за сутки до индукции артрита и продолжали ежедневно до истечения 6 суток после индукции. Результаты представлены в таблице 2.
| Таблица 2 |
| |
% снижения AUC (по сравнению с контролем-носителем для артрита) |
| Опухоль голеностопного сустава |
Порог вон Фрея |
| Р2Х7-антагонист 1 |
2,6±11,6 |
26,5±11,4 |
| Рофекоксиб |
50,6±4,7** |
29,8±7,8* |
| Р2Х7-антагонист 1 + рофекоксиб |
56,1±6,4** |
69,5±6,6** тестирование взаимодействия р=0,44*** |
| * р<0,05, ** р<0,0001 по сравнению с контролем-носителем для артрита, |
| *** величина взаимодействия, указывающая на аддитивный эффект комбинации. |
Из вышеуказанных результатов можно видеть, что комбинация Р2Х7-антагониста 1 и рофекоксиба показала позитивное взаимодействие, проявлявшееся в снижении механического порога. Обнаружение того, что эти два лекарства оказывают позитивный эффект в отношении порога вон Фрея в комбинации, которая показывает незначительное благоприятное действие в отношении опухоли голеностопного сустава, указывает на то, что эта комбинация лекарств обладает сильным и неожиданно позитивным эффектом в отношении боли в воспаленном суставе. Кроме того, анализ образцов крови из этих исследований продемонстрировал, что фармакокинетические профили двух лекарств при их введении в комбинации были идентичны профилям при их индивидуальном введении. Это указывает на то, что обнаруженные позитивные эффекты не могут быть приписаны изменениям в фармакокинетических профилях лекарств, а являются результатом фармакологического взаимодействия.
Пример 3В
Оценка противовоспалительной активности комбинаций ингибитор СОХ-2, вальдекоксиб/Р2Х7-антагонист в модели артрита, индуцированного у крыс клеточной стенкой стрептококка1
Противовоспалительную активность ингибитора СОХ-2, вальдекоксиба, в комбинации с Р2Х7-антагонистом оценивали, используя протокол, описанный в примере 3А. Р2Х7-антагонист 1 вводили перорально в дозе 30 мг/кг (4 мл/кг, два раза в сутки) в виде суспензии в 1%-ной (мас./об.) метилцеллюлозе в деионизированной воде вместе с вальдекоксибом (Pfizer) (1 мг/кг) в едином препарате. Введение доз начинали за сутки до индукции артрита и продолжали ежедневно до истечения 6 суток после индукции. Результаты представлены в таблице 3.
| Таблица 3 |
| |
% снижения AUC (по сравнению с контролем-носителем для артрита) |
| Опухоль голеностопного сустава |
Порог вон Фрея |
| Р2Х7-антагонист 1 |
2,6±11,6 |
26,5±11,4 |
| вальдекоксиб |
52,8±3,1** |
37,8±8,6* |
| Р2Х7-антагонист 1 + вальдекоксиб |
57,4±6,8** |
60,9±6,0** тестирование взаимодействия р=0,85*** |
| * р<0,01, ** р<0,0001 по сравнению с контролем-носителем для артрита, |
| *** величина взаимодействия, указывающая на аддитивный эффект комбинации. |
Из вышеуказанных результатов можно видеть, что комбинация Р2Х7-антагониста 1 и вальдекоксиба показала позитивное взаимодействие, проявлявшееся в снижении механического порога. Обнаружение того, что эти два лекарства оказывают позитивный эффект в отношении порога вон Фрея в комбинации, которая показывает незначительное благоприятное действие в отношении опухоли голеностопного сустава, указывает на то, что эта комбинация лекарств обладает сильным и неожиданно позитивным эффектом в отношении боли в воспаленном суставе. Кроме того, анализ образцов крови из этих исследований продемонстрировал, что фармакокинетические профили двух лекарств при их введении в комбинации были идентичны профилям при их индивидуальном введении. Это указывает на то, что обнаруженные позитивные эффекты не могут быть приписаны изменениям в фармакокинетических профилях лекарств, а являются результатом фармакологического взаимодействия.
Формула изобретения
1. Фармацевтическая композиция для лечения артритов, содержащая в смеси первый активный ингредиент, который представляет собой антагонист Р2Х7-рецептора, и второй активный ингредиент, выбранный из целекоксиба, рофекоксиба и вальдекоксиба, где антагонист Р2Х7-рецептора представляет собой производное адамантила формулы
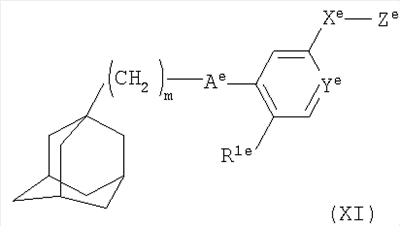
где m равно 1, 2 или 3;
Ае представляет собой C(O)NH или NHC(O);
Ye представляет собой N или СН;
Xе представляет собой связь, СО, (CH2)1-6, O(CH2)1-6, (CH2)1-6NH(CH2)1-6, (CH2)1-6O(CH2)1-6, NH(CH2)1-6;
Ze представляет собой NR2eR3e;
R1e представляет собой галоген, циано, нитро, амино, гидроксил, C1-C6алкил или С3-С8циклоалкил, причем алкильная или циклоалкильная группа может быть возможно замещена одним или более атомами фтора;
каждый из R2e и R3e независимо представляет собой атом водорода, C1-С6алкил или С3-С8циклоалкил, причем алкильная или циклоалкильная группа может быть возможно замещена одной или более группами, выбранными из гидроксила, галогена или С1-С6алкокси;
или R2e и R3e вместе с атомом азота, к которому они присоединены, образуют 3-9-членное насыщенное моно- или бициклическое гетероциклическое кольцо, содержащее от 1 до 2 атомов азота и возможно атом кислорода, которое может быть возможно замещено одной или более группами, выбранными из гидроксила, галогена или С1-С6алкокси; или его фармацевтически приемлемую соль или сольват.
2. Композиция по п.1, где антагонист Р2Х7-рецептора представляет собой
2-хлор-5-[[2-(2-гидрокси-этиламино)-этиламино]-метил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-[3-[(3-гидроксипропил)амино]пропил]-N-(трицикло[3.3.1.1]дец-1-илметил)-бензамид,
(R)-2-хлор-5-[3-[(2-гидрокси-1-метилэтил)амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-[[2-[(2-гидроксиэтил)амино]этокси]метил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-[3-[3-(метиламино)пропокси]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)бензамид,
2-хлор-5-[3-(3-гидрокси-пропиламино)-пропокси]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-[2-(3-гидроксипропиламино)этиламино]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-пиперазин-1-илметил-N-(трицикло[3.3.1.1]дец-1-илметил)-бензамид,
2-хлор-5-(2,5-диазабицикло[2.2.1]гепт-2-илметил)-N-(трицикпо[3.3.1.1]дец-1-илметил)-бензамид,
5-хлор-2-[3-[(3-гидроксипропил)амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамид,
5-хлор-2-[3-(этиламино)пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамид,
5-хлор-2-[3-[(2-гидроксиэтил)амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамид,
5-хлор-2-[3-[[(2S)-2-гидроксипропил)амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамид,
N-[2-метил-5-(9-окса-3,7-диазабицикло[3.3.1]нон-3-илкарбонил)фенил]трицикло[3.3.1.13,7]декан-1-ацетамид,
или его фармацевтически приемлемую соль или сольват.
3. Композиция по п.1 или 2, где второй активный ингредиент представляет собой целекоксиб.
4. Композиция по п.1 или 2, где второй активный ингредиент представляет собой рофекоксиб.
5. Композиция по п.1 или 2, где второй активный ингредиент представляет собой вальдекоксиб.
6. Композиция по п.1 или 2, которая приготовлена для перорального введения.
7. Способ получения фармацевтической композиции по любому из пп.1-6, включающий смешивание первого активного ингредиента со вторым активным ингредиентом.
8. Применение композиции по любому из пп.1-6 в изготовлении лекарства для лечения артритов.
9. Применение по п.8, где артрит представляет собой ревматоидный артрит или остеоартрит.
10. Способ лечения артритов, включающий введение терапевтически эффективного количества фармацевтической композиции по любому из пп.1-6 нуждающемуся в этом пациенту.
11. Способ по п.10, где артрит представляет собой ревматоидный артрит или остеоартрит.
12. Фармацевтический продукт для лечения артритов, содержащий в комбинации препарат первого активного ингредиента, который представляет собой антагонист Р2Х7-рецептора, и препарат второго активного ингредиента, выбранного из целекоксиба, рофекоксиба или вальдекоксиба, для одновременного, последовательного или раздельного использования в терапии, где антагонист Р2Х7-рецептора представляет собой производное адамантила формулы (XI), как определено в п.1.
13. Набор для лечения артритов, содержащий препарат первого активного ингредиента, который представляет собой антагонист Р2Х7-рецептора, препарат второго активного ингредиента, выбранного из целекоксиба, рофекоксиба или вальдекоксиба, и инструкции по одновременному, последовательному или раздельному введению этих препаратов, нуждающемуся в этом пациенту, где антагонист Р2Х7-рецептора представляет собой производное адамантила формулы (XI), как определено в п.1. |
|