(54) ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ АНТАГОНИСТ Р2Х7 РЕЦЕПТОРА И ФАКТОР НЕКРОЗА ОПУХОЛИ 
(57) Реферат:
Заявленное изобретение относится к химико-фармацевтической промышленности и касается фармацевтического продукта для лечения артрита, содержащего соединение (XI), представляющее собой антагонист Р2Х7 рецептора, причем указанный антагонист Р2Х7 рецептора представляет собой производное адамантила, и второй активный ингредиент, который представляет собой ингибитор фактора некроза опухоли (ФНО). Заявленное изобретение обеспечивает четко выраженный благоприятный противовоспалительный эффект и ослабление боли. 4 н. и 4 з.п. ф-лы, 1 табл.

Настоящее изобретение относится к комбинациям фармацевтически активных веществ для применения в лечении воспалительных состояний/расстройств, особенно ревматоидного артрита.
Хронические воспалительные расстройства, такие как ревматоидный артрит, являются полигенными, высококомплексными и затрагивают разнообразные воспалительные и иммунные механизмы. Лечение этих расстройств в значительной степени опирается на опыт применения множества терапевтических агентов с малоизученными вовлекаемыми механизмами. Недавнее исследование предполагает, что два воспалительных медиатора, цитокины IL-1 (интерлейкин-1) и ФНОальфа (ФНО), могут играть ключевую роль в воспалительном процессе при ревматоидном артрите.
Было бы желательно разработать новые фармацевтические препараты для применения в лечении воспалительных состояний/расстройств.
Согласно настоящему изобретению предложен фармацевтический продукт, содержащий в сочетании препарат первого активного ингредиента, который представляет собой антагонист Р2Х7 рецептора, причем антагонист Р2Х7 рецептора является производным адамантила, и препарат второго активного ингредиента, который представляет собой ингибитор фактора некроза опухоли (ФНО), для одновременного, последовательного или раздельного применения в терапии.
В другом аспекте согласно изобретению предложен набор, содержащий препарат первого активного ингредиента, который представляет собой антагонист Р2Х7 рецептора, причем антагонист Р2Х7 рецептора является производным адамантила, препарат второго активного ингредиента, который представляет собой ингибитор фактора некроза опухоли (ФНО), и инструкции для одновременного, последовательного или раздельного введения препаратов пациенту, нуждающемуся в этом.
Р2Х7 рецептор (ранее известный как P2Z рецептор) представляет собой лиганд-управляемый ионный канал, который присутствует во множестве клеточных типов, в значительной степени в тех, которые, как известно, вовлечены в воспалительный/иммунный процесс, а именно макрофагах, тучных клетках и лимфоцитах (Т и В). Активация Р2Х7 рецептора внеклеточными нуклеотидами, в частности аденозинтрифосфатом, как известно, приводит, среди прочего, к высвобождению интерлейкина-1 (IL-1). (IL-1).
Антагонист Р2Х7 рецептора представляет собой соединение или другое вещество, которое способно предупреждать либо полностью, либо частично активацию Р2Х7 рецептора.
Способы анализа антагонизма Р2Х7 рецептора известны в уровне техники, например, из WO 01/42194, в которой описан анализ, основанный на наблюдении того, что, когда Р2Х7 рецептор активируют при использовании агониста рецептора в присутствии бромида этидия (флуоресцентный ДНК-зонд), наблюдают увеличение флуоресценции связанного с внутриклеточной ДНК бромида этидия. Таким образом, увеличение флуоресценции может быть использовано в качестве меры активации Р2Х7 рецептора и, следовательно, для подсчета действия соединения или вещества на Р2Х7 рецептор.
В WO 01/42194 анализ проводят посредством использования 96-луночного микротитрационного планшета с плоским дном и заполнения лунок 250 мкл исследуемого раствора, содержащего 200 мкл суспензии ТНР-1 клеток (клеток человеческой острой моноцитарной лейкемии) (2,5×106 клеток/мл), которая содержит 10-4 М бромида этидия, 25 мкл буферного раствора с высокой концентрацией калия, содержащего 10-5 М бензоилбензоиладенозинтрифосфата (bbATP, известного агониста Р2Х7 рецептора), и 25 мкл буферного раствора с высокой концентрацией калия, содержащего 3×10-5 М исследуемого соединения. Планшет накрывают листом пластика и инкубируют при 37°С в течение 1 часа. Планшет затем считывают в спектрофлуориметре для планшетов Perkin-Elmer, возбуждение 520 нм, эмиссия 595 нм, ширина щели: для возбуждения (Ех) 15 нм, для эмиссии (Em) 20 нм. С целью сравнения, bbATP (агонист Р2Х7 рецептора) и пиридоксаль-5-фосфат (антагонист Р2Х7 рецептора) используют в тесте отдельно в качестве контролей. Из полученных показаний прибора рассчитывают значение pIC50 для исследуемого соединения, и это значение представляет собой кологарифм концентрации исследуемого соединения, которая необходима, чтобы снизить активность агониста bbATP на 50%. Значение pIC50 больше 5,5 является нормальным признаком антагониста.
Примеры антагонистов Р2Х7 рецептора, которые могут быть использованы в соответствии с настоящим изобретением, включают соединения, описанные в WO 00/61569, WO 01/42194, WO 01/44170 и WO 03/41707, полное содержание которых дано здесь посредством ссылки.
Более конкретно, в первом воплощении настоящего изобретения антагонист Р2Х7 рецептора представляет собой соединение формулы
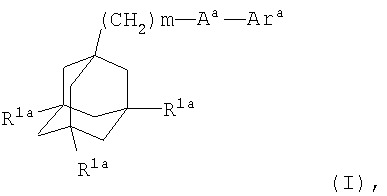
где m представляет собой 1, 2 или 3;
каждый R1a независимо представляет собой атом водорода или галогена;
Аa представляет собой C(O)NH или NHC(О);
Ara представляет собой группу
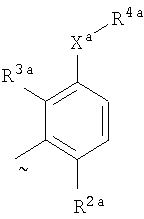 или или 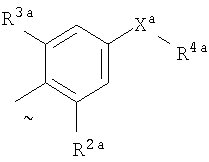
Xa представляет собой связь, атом кислорода или группу СО, (CH2)1-6, СН=, (CH2)1-6O, O(CH2)1-6, O(СН2)2-6О, O(СН2)2-3О(СН2)1-3, CR'(OH), (CH2)1-3O(СН2)1-3 (СН2)1-3O(СН2)2-3O, NR5a, (CH2)1-6NR5а, NR5а(CH2)1-6, (CH2)1-3NR5a(CH2)1-3, О(CH2)2-6NR5a, О(CH2)2-3NR5a(CH2)1-3, (CH2)1-3NR5a(CH2)2-3O, NR5a(CH2)2-6O, NR5a(CH2)2-3O(CH2)1-3, CONR5а, NR5аCO, S(О)n, S(О)nCH2, CH2S(О)n, SO2NR5a или NR5aSO2;
n представляет собой 0, 1 или 2;
R' представляет собой атом водорода или С1-С6алкильную группу;
один из R2a и R3a представляет собой галоген, циано, нитро, амино, гидроксил или группу, выбранную из (1) С1-С6алкила, возможно замещенного по меньшей мере одним С3-С6циклоалкилом, (2) С3-С8циклоалкила, (3) С1-С6алкилокси, возможно замещенного по меньшей мере одним С3-С6циклоалкилом, и (4) С3-С8циклоалкилокси, причем каждая из этих групп возможно замещена одним или более чем одним атомом фтора, а другой из R2а и R3а представляет собой атом водорода или галогена;
либо R4а представляет собой 3-9-членную насыщенную или ненасыщенную алифатическую гетероциклическую кольцевую систему, содержащую один или два атома азота и возможно атом кислорода, причем гетероциклическая кольцевая система возможно замещена одним или более чем одним заместителем, независимо выбранным из атомов фтора, гидроксила, карбоксила, циано, С1-С6алкила, С1-С6гидроксиалкила, -NR6аR7а, -(CH2)rNR6aR7a и -CONR6aR7a,
либо R4a представляет собой 3-8-членную насыщенную карбоциклическую кольцевую систему, замещенную одним или более чем одним заместителем, независимо выбранным из атома -NR6aR7a, -(CH2)rNR6aR7a и -CONR6aR7a, причем кольцевая система возможно дополнительно замещена одним или более чем одним заместителем, независимо выбранным из атомов фтора, гидроксила и C1-С6алкила;
r представляет собой 1, 2, 3, 4, 5 или 6;
R5a представляет собой атом водорода или С1-С6алкильную или С1-С6ациклоалкильную группу;
R6а и R7а каждый независимо представляет собой атом водорода или С1-С6алкильную, С2-С6гидроксиалкильную или С2-С8циклоалкильную группу, или R6а и R7а вместе с атомом азота, к которому они присоединены, образуют 3-8-членное насыщенное гетероциклическое кольцо;
при условии, что
а) когда Аa представляет собой С(O)NH и R4a представляет собой незамещенную 3-8-членную насыщенную алифатическую гетероциклическую кольцевую систему, содержащую один атом азота, тогда Xa не является связью, и
б) когда Аa представляет собой С(O)NH и Xa представляет собой группу (СН2)1-6 или O(СН2)1-6, тогда R4a не является незамещенной имидазолильной, незамещенной морфолинильной, незамещенной пиперидинильной или незамещенной пирролидинильной группой, и
в) когда Аa представляет собой NHC(О) и R4a представляет собой незамещенную 3-8-членную насыщенную алифатическую гетероциклическую кольцевую систему, содержащую один атом азота, тогда Xa не является связью, и
г) когда Аa представляет собой NHC(O) и Xa представляет собой O(CH2)1-6, NH(CH2)1-6 или SCH2, тогда R4a не является незамещенной 1-пиперидинильной или незамещенной 1-пирролидинильной группой, и
д) когда Aa представляет собой NHC(O) и Хa представляет собой O(СН2)2-3NH(СН2)2, тогда R4a не является имидазолильной группой;
или его фармацевтически приемлемые соль или сольват.
Соединения формулы (I) описаны в WO 00/61569.
Во втором воплощении настоящего изобретения антагонист Р2Х7 рецептора представляет собой соединение формулы
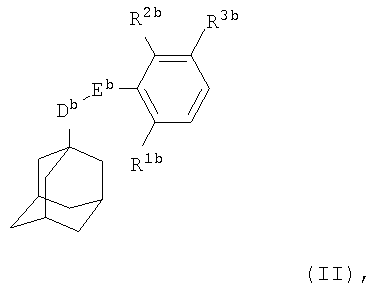
где Db представляет собой CH2 или СН2СН2;
Еb представляет собой C(O)NH или NHC(О);
R1b и R2b каждый независимо представляет собой атом водорода или галогена или группу амино, нитро, C1-С6алкильную или трифторметильную группу;
R3b представляет собой группу формулы
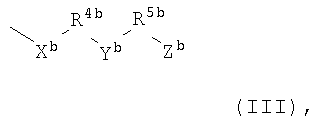
Хb представляет собой атом кислорода или серы или группу NH, SO или SO2;
Yb представляет собой атом кислорода или серы или группу NR11b, SO или SO2;
Zb представляет собой группу -ОН, -SH, -CO2Н, С1-С6алкокси, С1-С6алкилтио, С1-С6алкилсульфинил, С1-С6алкилсульфонил, -NR6bR7b, -C(O)NR8bR9b, имидазолил, 1-метилимидазолил, -N(R10b)C(O)-C1-C6алкил, С1-С6алкилкарбонилокси, С1-С6алкоксикарбонилокси, -OC(О)NR12bR13b, -OCH2OC(O)R14b, -OCHOC(O)OR15b или -OC(O)OCH2OR16b;
R4b представляет собой С2-С6алкильную группу;
R5b представляет собой С1-С6алкильную группу;
R6b, R7b, R8b, R9b, R10b, R12b и R13b каждый независимо представляет собой атом водорода или С1-С6алкильную группу, возможно замещенную по меньшей мере одной гидроксильной группой;
R11b представляет собой атом водорода или С1-С6алкильную группу, возможно замещенную по меньшей мере одним заместителем, независимо выбранным из гидроксила и С1-С6алкокси; и
R14b, R15b и R16b каждый независимо представляет собой С1-С6алкильную группу;
при условии, что (1) когда Еb представляет собой NHC(О), Хb представляет собой О, S или NH, и Yb представляет собой О, тогда Zb представляет собой группу -NR6bR7b, где R6b представляет собой атом водорода, a R7b представляет собой либо атом водорода, либо C С1-С6алкильную группу, замещенную по меньшей мере одной гидроксильной группой, и (2) когда Eb представляет собой NHC(O), Xb представляет собой О, S или NH, Yb представляет собой NH, и R5b представляет собой СН2СН2, тогда Zb не является -ОН или имидазолилом;
или его фармацевтически приемлемые соль или сольват.
Соединения формулы (II) описаны в WO 01/42194.
В третьем воплощении настоящего изобретения антагонист Р2Х7 рецептора представляет собой соединение формулы
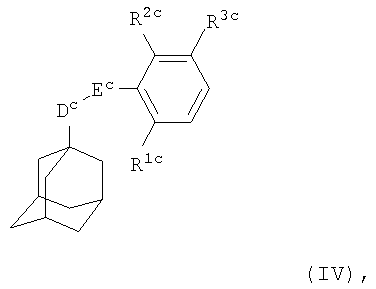
где Dc представляет собой СН2 или СН2СН2;
Еc представляет собой C(O)NH или NHC(O);
R1c и R2c каждый независимо представляет собой водород, галоген, амино, нитро, С1-С6алкил или трифторметил, но R1c и R2c не могут оба одновременно представлять собой водород;
R3c представляет собой группу формулы
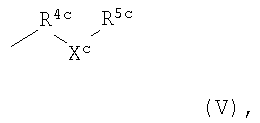
R4c представляет собой С1-С6алкильную группу;
Xc представляет собой атом кислорода или серы или группу NR13c, SO или SO2;
R5c представляет собой водород, или R50 представляет собой С1-С6алкил или С2-С6алкенил, каждый из которых может быть возможно замещен по меньшей мере одним заместителем, выбранным из галогена, гидроксила, (ди)-С1-С6-алкиламино, -YcR6c,
5- или 6-членное гетероароматическое кольцо, содержащее от 1 до 4 гетероатомов, независимо выбранных из азота, кислорода и серы, причем само гетероароматическое кольцо может быть возможно замещено по меньшей мере одним заместителем, выбранным из галогена, гидроксила и С1-С6алкила;
Yс представляет собой атом кислорода или серы или группу NH, SO или SO2;
R6с представляет собой группу -R7сZc, где R7c представляет собой С2-С6алкильную группу, а Zc представляет собой -ОН, -CO2Н, -NR8cR9c, -C(O)NR10cR11c или -N(R12c)С(O)-С1-С6алкильную группу, и в случае, где Yc представляет собой атом кислорода или серы или группу NH, R6с дополнительно представляет собой водород, С1-С6алкил, С1-С6алкилкарбонил, С1-С6алкоксикарбонил, -C(O)NR14cR15c, -CH2OC(O)R16c, -CH2OC(O)OR17c или -C(O)OCH2OR18c;
R8c, R9c, R10c, R11c и R12c каждый независимо представляет собой атом водорода или C1-С6алкильную группу;
R13c представляет собой водород, С3-С8циклоалкил, С3-С8циклоалкилметил или R13с представляет собой C1-С6алкильную группу, возможно замещенную по меньшей мере одним заместителем, выбранным из гидроксила и С1-С6алкокси; и
R14c, R15c, R16c, R17c и R18c каждый независимо представляет собой C1-С6алкильную группу;
при условии, что когда Еc представляет собой С(O)NH, Xc представляет собой О, NH или N(С1-С6алкил), тогда R5c не является атомом водорода или незамещенной С1-С6алкильной группой;
или его фармацевтически приемлемые соль или сольват.
Предпочтительными соединениями формулы (IV) являются соединения, где R5c представляет собой возможно замещенную С1-С6алкильную группу, причем предпочтительный заместитель представляет собой -Yc-R6c. Когда R5c замещен 5- или 6-членным гетероароматическим кольцом, содержащим от 1 до 4 гетероатомов, предпочтительно, чтобы число гетероатомов в кольце не превышало 2.
Соединения формулы (IV) описаны в WO 01/44170.
В четвертом воплощении настоящего изобретения антагонист Р2Х7 рецептора представляет собой соединение формулы
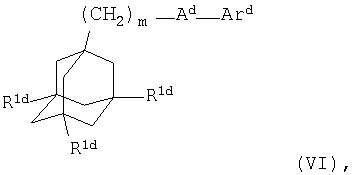
где m представляет собой 1, 2 или 3;
каждый R1d независимо представляет собой атом водорода или галогена;
Аd представляет собой C(O)NH или NHC(О);
Ard представляет собой группу
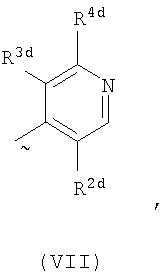 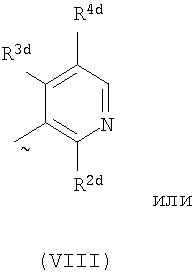 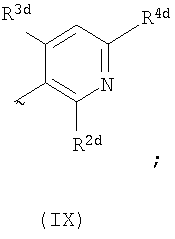
один из R2d и R3d представляет собой галоген, нитро, амино, гидроксил или группу, выбранную из (1) C1-С6алкила, возможно замещенного по меньшей мере одним атомом галогена, (2) C3-С8циклоалкила, (3) C1-С6алкокси, возможно замещенного по меньшей мере одним атомом галогена, и (4) C3-С8циклоалкилокси, а другой из R2b и R3b представляет собой атом водорода или галогена;
R4d представляет собой группу
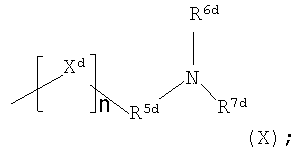
Xd представляет собой атом кислорода или серы или группу >N-R8d;
n представляет собой 0 или 1;
R5d представляет собой С1-С5алкильную группу, которая может быть возможно замещена по меньшей мере одним заместителем, выбранным из гидроксила, галогена и С1-С6алкокси;
R6d и R7d каждый независимо представляет собой атом водорода, С1-С6алкил (возможно замещенный по меньшей мере одним заместителем, выбранным из гидроксила, галогена, С1-С6алкокси и (ди)-С1-С4алкиламино (который сам возможно замещен по меньшей мере одной гидроксильной группой)) или С3-С8циклоалкил (возможно замещенный по меньшей мере одним заместителем, выбранным из гидроксила, галогена и С1-С6алкокси); и
R8d представляет собой атом водорода или С1-С5алкильную группу, которая может быть возможно замещена по меньшей мере одним заместителем, выбранным из гидроксила, галогена и С1-С6алкокси;
при условии, что:
а) когда n представляет собой 0, тогда Аd представляет собой NHC(O), и
б) когда n представляет собой 1, Xd представляет собой кислород, и Аd представляет собой C(O)NH, тогда R6d и R7d оба одновременно не являются атомом водорода или оба одновременно не являются незамещенным С1-С6алкилом, или когда один из R6d и R7d представляет собой атом водорода, тогда другой из R6d и R7d не представляет собой незамещенный С1-С6алкил, и
в) когда n представляет собой 1, Xd представляет собой кислород, серу или >NH, и Аd представляет собой NHC(O), тогда R6d и R7d оба одновременно не являются атомом водорода или оба одновременно не являются незамещенным С1-С6алкилом, или когда один из R6d и R7d представляет собой атом водорода, тогда другой из R6d и R7d не представляет собой незамещенный С1-С6алкил или -CH2CH2OH;
или его фармацевтически приемлемые соль или сольват.
Соединения формулы (VI) описаны в WO 03/41707.
В другом аспекте настоящего изобретения антагонист Р2Х7 рецептора представляет собой соединение формулы
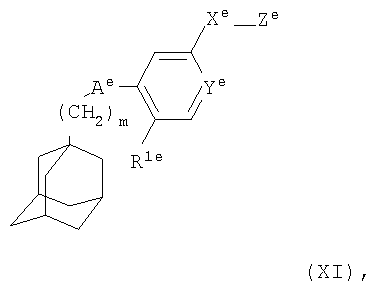
где m представляет собой 1, 2 или 3;
Аe представляет собой C(O)NH или NHC(O);
Ye представляет собой N или CH;
Xе представляет собой связь, СО, (CH2)1-6, O(CH2)1-6, (CH2)1-6NH(CH2)1-6, (CH2)1-6O(CH2)1-6, NH(CH2)1-6;
Zе представляет собой NR2eR3e;
R1e представляет собой галоген, циано, нитро, амино, гидроксил, C1-С6алкил или C3-С8циклоалкил, причем алкильная или циклоалкильная группа может быть возможно замещена одним или более чем одним атомом фтора;
R2e и R3e каждый независимо представляет собой атом водорода, C1-С6алкил или C3-С8циклоалкил, причем алкильная или циклоалкильная группа может быть возможно замещена одной или более чем одной группой, выбранной из гидроксила, галогена или C1-С6алкокси,
или R2e и R3e вместе с атомом азота, к которому они присоединены, образуют 3-9-членное насыщенное моно- или бициклическое гетероциклическое кольцо, содержащее от 1 до 2 атомов азота и возможно атом кислорода, причем гетероциклическое кольцо может быть возможно замещено одной или более чем одной группой, выбранной из гидроксила, галогена или C1-С6алкокси;
или его фармацевтически приемлемые соль или сольват.
Соединения формулы (XI) могут быть получены химическим способом согласно или аналогично способу, описанному в ссылках, упомянутых здесь выше.
В другом аспекте настоящего изобретения антагонист Р2Х7 рецептора представляет собой:
2-хлор-5-[[2-(2-гидрокси-этиламино)-этиламино]-метил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-[3-[(3-гидроксипропил)амино]пропил]-N-(трицикло[3.3.1.1]дец-1-илметил)-бензамид,
(R)-2-хлор-5-[3-[(2-гидрокси-1-метилэтил)амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-[[2-[(2-гидроксиэтил)амино]этокси]метил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-[3-[3-(метиламино)пропокси]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-[3-(3-гидрокси-пропиламино)-пропокси]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-[2-(3-гидроксипропиламино)этиламино]-N-(трицикло[3.3.1.13,7]дец-1-илметил)бензамид,
2-хлор-5-[2-(3-гидроксипропилсульфонил)этокси]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-[2-[2-[(2-гидроксиэтил)амино]этокси]этокси]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-[[2-[[2-(1-метил-1H-имидазол-4-ил)этил]амино]этил]амино]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-пиперазин-1-илметил-N-(трицикло[3.3.1.1]дец-1-илметил)-бензамид,
2-хлор-5-(4-пиперидинилокси)-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-(2,5-диазабицикло[2.2.1]гепт-2-илметил)-N-(трицикло[3.3.1.1]дец-1-илметил)-бензамид,
2-хлор-5-(пиперидин-4-илсульфинил)-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
5-хлор-2-[3-[(3-гидроксипропил)амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамид,
2-хлор-5-[3-[[(1R)-2-гидрокси-1-метилэтил]амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-3-пиридинкарбоксамид,
5-хлор-2-[3-(этиламино)пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамид,
5-хлор-2-[3-[(2-гидроксиэтил)амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамид,
5-хлор-2-[3-[[(2S)-2-гидроксипропил]амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамид,
N-[2-метил-5-(9-окса-3,7-диазабицикло[3.3.1]нон-3-илкарбонил)фенил]-трицикло[3.3.1.13,7]декан-1-ацетамид
или фармацевтически приемлемые соль или сольват любого из них.
Фармацевтически приемлемые соли включают там, где применимо, соли присоединения кислот, производные от фармацевтически приемлемых неорганических и органических кислот, такие как хлорид, бромид, сульфат, фосфат, малеат, фумарат, тартрат, цитрат, бензоат, 4-метоксибензоат, 2- или 4-гидроксибензоат, 4-хлорбензоат, пара-толуолсульфонат, метансульфонат, аскорбат, ацетат, сукцинат, лактат, глутарат, глюконат, трикарбаллилат, гидроксинафталинкарбоксилат или олеат; и соли, полученные из фармацевтически приемлемых неорганических и органических оснований. Соли, производные от неорганических оснований, включают соли алюминия, аммония, кальция, меди, железа (III), железа (II), лития, магния, марганца (III), марганца (II), калия, натрия, цинка и висмута. Особенно предпочтительными являются соли аммония, кальция, магния, калия и натрия. Соли, производные от фармацевтически приемлемых органических основания, включают соли первичных, вторичных и третичных аминов, циклических аминов, подобных аргинину, бетаину, холину и тому подобное. Примеры фармацевтически приемлемых сольватов включают гидраты.
Примеры антагонистов Р2Х7 рецептора, которые могут быть использованы в настоящем изобретении, включают:
дигидрохлорид 2-хлор-5-[[2-(2-гидрокси-этиламино)-этиламино]-метил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамида,
гидрохлорид 2-хлор-5-[3-[(3-гидроксипропил)амино]пропил]-N-(трицикло[3.3.1.1]дец-1-илметил)-бензамида,
гидрохлорид (R)-2-хлор-5-[3-[(2-гидрокси-1-метилэтил)амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамида,
ацетат (1:1) 2-хлор-5-([2-[(2-гидроксиэтил)амино]этокси]метил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамида,
гидрохлорид 2-хлор-5-[3-[3-(метиламино)пропокси]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамида,
гидрохлорид 2-хлор-5-[3-(3-гидрокси-пропиламино)-пропокси]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамида,
ацетат (1:1) 2-хлор-5-[2-(3-гидроксипропиламино)этиламино]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамида,
2-хлор-5-[2-(3-гидроксипропилсульфонил)этокси]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
гидрохлорид 2-хлор-5-[2-[2-[(2-гидроксиэтил)амино]этокси]этокси]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамида,
2-хлор-5-[[2-[[2-(1-метил-1H-имидазол-4-ил)этил]амино]этил]амино]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
дигидрохлорид 2-хлор-5-пиперазин-1-илметил-N-(трицикло[3.3.1.1]дец-1-илметил)-бензамида,
гидрохлорид 2-хлор-5-(4-пиперидинилокси)-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамида,
гидрохлорид 2-хлор-5-(2,5-диазабицикло[2.2.1]гепт-2-илметил)-N-(трицикло[3.3.1.1]дец-1-илметил)-бензамида,
2-хлор-5-(пиперидин-4-илсульфинил)-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
5-хлор-2-[3-[(3-гидроксипропил)амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамид,
2-хлор-5-[3-[[(1R)-2-гидрокси-1 -метилэтил]амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-3-пиридинкарбоксамид,
гидрохлорид 5-хлор-2-[3-(этиламино)пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамида,
гидрохлорид 5-хлор-2-[3-[(2-гидроксиэтил)амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамида,
дигидрохлорид 5-хлор-2-[3-[[(23)-2-гидроксипропил]амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-пиридинкарбоксамида и
гидрохлорид N-[2-метил-5-(9-окса-3,7-диазабицикло[3.3.1]нон-3-илкарбонил)фенил]-трицикло[3.3.1.13,7]декан-1-ацетамида.
Антагонист Р2Х7 рецептора, используемый в настоящем изобретении, может существовать в стереоизомерных формах. Следует понимать, что изобретение включает все геометрические и оптические изомеры активного ингредиента и их смеси, включая рацематы. Таутомеры и их смеси также составляют аспект настоящего изобретения.
Второй активный ингредиент в продукте или наборе по настоящему изобретению представляет собой ингибитор фактора некроза опухоли (ФНО). Ингибитор ФНО представляет собой соединение или другое вещество, которое способно ингибировать активность ФНО либо полностью, либо частично. Подробное описание соединений или веществ, которые могут быть использованы в настоящем изобретении в качестве ингибиторов ФНО, можно найти, например, в опубликованной международной заявке на патент № WO 98/05357, полное содержание которой включено сюда посредством ссылки.
В одном воплощении изобретения ингибитор фактора некроза опухоли (ФНО) представляет собой рецепторную молекулу, способную связываться с ФНО. Такие рецепторные молекулы известны в уровне техники, а подробное описание рецепторных молекул, которые могут быть использованы в настоящем изобретении, можно найти, например, на страницах с 32 по 35 WO 98/05357. Примером рецепторной молекулы, которая дает особенно хорошие результаты в настоящем изобретении, является этанерсепт (Expert Орт. Pharmacother. (2001) 2(7); 1137-1148). Этанерсепт представляет собой слитый белок, состоящий из внеклеточного лиганд-связывающего участка рецептора фактора некроза опухоли человека, связанный с Fc-участком IgG (иммуноглобулина G) человека. Его получают с помощью технологии рекомбинантных ДНК в системе экспрессии в клетках млекопитающих СНО (клетки яичников китайского хомячка). Считают, что этанерсепт ослабляет биологическую активность провоспалительного цитокина - фактора некроза опухоли (ФНО) посредством связывания белка и блокирования его взаимодействия с клеточными поверхностными рецепторами ФНО. Этанерсепт продается Amgen and Wyeth Pharmaceuticals под торговым наименованием "Энбрел". Другим примером ингибитора ФНО, который представляет собой рецепторную молекулу и который может быть использован согласно настоящему изобретению, является пегсунерсепт (Pegsunercept), пегилированный ингибитор растворимого рецептора ФНО (Arth. Rheum. (2003) 48, S121).
В другом воплощении изобретения ингибитор фактора некроза опухоли (ФНО) представляет собой антитело против ФНО. Примеры антител против ФНО согласно настоящему изобретению включают моноклональные, химерные, гуманизированные, поверхностные (resurfaced) и рекомбинантные антитела и их фрагменты, которые способны ингибировать активность ФНО либо полностью, либо частично. Такие антитела известны в уровне техники и описаны, например, на страницах с 13 по 32 WO 98/05357. Конкретными примерами антител против ФНО, которые могут быть использованы в настоящем изобретении, являются моноклональные антитела инфликсимаб и адалимумаб (D2E7). Инфликсимаб представляет собой химерное моноклональное антитело IgG1k, состоящее из человеческого константного и мышиного вариабельного участков и продается Centocor под торговым наименованием "Ремикейд". Адалимумаб (D2E7) представляет собой рекомбинантное человеческое IgG1 моноклональное антитело, полученное при использовании технологии фагового дисплея, приводящей к получению антитела с вариабельными участками тяжелых и легких цепей человеческого происхождения и константными участками человеческого IgG1k. Адалимумаб (D2E7) продается Abbott Laboratories под торговым наименованием "Хумира". Другим примером фрагмента антитела против ФНО, который может быть использован согласно настоящему изобретению, является CDP-870, фрагмент пегилированного гуманизированного антитела, который с высоким сродством связывается с ФНО (Cur. Opin. Investig. Drugs. (2003) 4; 588-592).
В другом воплощении изобретения ингибитор фактора некроза опухоли (ФНО) может связывать рецептор ФНО и включает антитела против рецептора ФНО.
Обнаружено, что выбор активных ингредиентов по изобретению является выгодным, поскольку это приводит к целебному противовоспалительному действию и, соответственно, может быть использован для лечения различных острых и хронических воспалительных состояний/расстройств, таких как ревматоидный артрит. Лечение воспалительных расстройств может включать уменьшение опухоли и/или облегчение боли, ассоциированные с этим состоянием. В этом отношении продукты по настоящему изобретению оказались особенно полезны при уменьшении или облегчении боли, вызванной воспалительными расстройствами, в частности ревматоидным артритом.
Другой полезный аспект настоящего изобретения состоит в том, что оно может обеспечить эффективное лечение с использованием более низких доз ингибитора ФНО, чем это возможно при использовании только ингибитора ФНО. Это существенно, поскольку использование биологических терапевтических агентов, таких как ингибиторы ФНО, может делать пациентов восприимчивыми к инфекциям, вызванным условно-патогенными микроорганизмами. Более того, общепринятая анти-ФНО терапия, такая как терапия этанерсептом, имеет дополнительную трудность, состоящую в том, что он имеет длительный период "выведения", до того как лекарство удаляется из организма. Совместное введение с Р2Х7 антагонистом, которое позволяет снизить дозу ингибитора ФНО, не подвергая риску эффективность, уменьшает проблемы, связанные с безопасностью, и потенциально позволяет применять анти-ФНО терапию для пациентов там, где ее применение до сих пор считается нецелесообразным.
В предпочтительном воплощении настоящего изобретения второй активный ингредиент представляет собой ингибитор фактора некроза опухоли (ФНО) этанерсепт, а первый активный ингредиент, который представляет собой антагонист Р2Х7 рецептора, выбран из
2-хлор-5-[[2-(2-гидроксиэтиламино)-этиламино]метил]-N-(трицикпо[3.3.1.13,7]дец-1-илметил)бензамида,
2-хлор-5-[3-[(3-гидроксипропил)амино]пропил]-N-(трицикло[3.3.1.1]дец-1-илметил)бензамида,
(R)-2-хлор-5-[3-[(2-гидрокси-1-метилэтил)амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)бензамида,
2-хлор-5-[[2-[(2-гидроксиэтил)амино]этокси]метил]-N-(трицикпо[3.3.1.13,7]дец-1-илметил)бензамида,
2-хлор-5-[3-[3-(метиламино)пропокси]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)бензамида,
2-хлор-5-[3-(3-гидроксипропиламино)пропокси]-N-(трицикло[3.3.1.13,7]дец-1-илметил)бензамида,
2-хлор-5-[2-(3-гидроксипропиламино)этиламино]-N-(трицикло[3.3.1.13,7]дец-1-илметил)бензамида,
2-хлор-5-[2-(3-гидроксипропилсульфонил)этокси]-N-(трицикло[3.3.1.13,7]дец-1-илметил)бензамида,
2-хлор-5-[2-[2-[(2-гидроксиэтил)амино]этокси]этокси]-N-(трицикло[3.3.1.13,7]дец-1-илметил)бензамида,
2-хлор-5-[[2-[[2-(1-метил-1Н-имидазол-4-ил)этил]амино]этил]амино]-N-(трицикло[3.3.1.13,7]дец-1-илметил)бензамида,
2-хлор-5-пиперазин-1-илметил-N-(трицикло[3.3.1.1]дец-1-илметил)бензамида,
2-хлор-5-(4-пиперидинилокси)-N-(трицикло[3.3.1.13,7]дец-1-илметил)бензамида,
2-хлор-5-(2,5-диазабицикло[2.2.1]гепт-2-илметил)-N-(трицикло[3.3.1.1]дец-1-илметил)бензамида,
2-хлор-5-(пиперидин-4-илсульфинил)-N-(трицикло[3.3.1.13,7]дец-1-илметил)бензамида,
5-хлор-2-[3-[(3-гидроксипропил)амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамида,
2-хлор-5-[3-[[(1R)-2-гидрокси-1-метилэтил]амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)3-пиридинкарбоксамида,
5-хлор-2-[3-(этиламино)пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамида,
5-хлор-2-[3-[(2-гидроксиэтил)амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамида,
5-хлор-2-[3-[[(2S)-2-гидроксипропил]амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамида,
N-[2-метил-5-(9-окса-3,7-диазабицикло[3.3.1]нон-3-илкарбонил)фенил]-трицикло[3.3.1.13,7]декан-1-ацетамида
или фармацевтически приемлемых соли или сольвата любого из них. Продукты этого воплощения, в частности, могут быть использованы для уменьшения или облегчения боли, вызванной воспалительными расстройствами, особенно ревматоидным артритом.
Первый и второй активные ингредиенты вводят одновременно (не в смеси), последовательно или раздельно для лечения воспалительных состояний. Последовательно означает, что первый и второй активные ингредиенты вводят в любом порядке, непосредственно один за другим. Они также обладают требуемым действием, если их вводят раздельно.
Первый и второй активные ингредиенты удобно вводить посредством перорального или парентерального (например, внутривенного, подкожного, внутримышечного, внутрисуставного или ингаляционного) введения, при использовании традиционных лекарственных форм системного действия, таких как таблетки, капсулы, пилюли, порошки, водные или масляные растворы или суспензии, эмульсии и стерильные инъекционные водные или масляные растворы или суспензии. Эти лекарственные формы будут обычно включать один или более чем один фармацевтически приемлемый ингредиент, который может быть выбран, например, из адъювантов, носителей, связывающих веществ, смазывающих веществ, разбавителей, стабилизирующих веществ, буферных веществ, эмульгаторов, веществ, регулирующих вязкость, сурфактантов, консервантов, корригентов и красителей. Как будет понятно специалистам в данной области техники, наиболее подходящий способ введения активных ингредиентов зависит от ряда факторов. Однако в основном пероральное введение первого активного ингредиента является предпочтительным, тогда как для второго активного ингредиента подкожное введение является предпочтительным.
Для вышеупомянутых терапевтических применений вводимые дозы, конечно, будут варьироваться в зависимости от используемых первого и второго активных ингредиентов, способа введения, требуемого лечения и указанного состояния или расстройства. Однако в основном удовлетворительные результаты могут быть получены, когда суммарная, объединенная доза первого и второго активных ингредиентов находится в пределах от 10 до 2000 миллиграмм (мг), в частности от 10, 20, 30, 40, 50, 100, 150, 200 или 300 до 1800, 1500, 1200, 1000, 800, 600, 500 или 400 мг.
Фармацевтический продукт или набор по изобретению можно вводить в виде разделенных доз. При введении путем разделенных доз первый и второй ингредиенты могут быть введены с различной частотой, независимо друг от друга. Однако в основном частота введения каждого из активных ингредиентов будет независимо находиться в пределах от одной дозы каждые 7 дней до 4 доз в день.
В одном воплощении настоящего изобретения доза первого активного ингредиента в фармацевтическом продукте или наборе находится в пределах от 5 до 1000 мг, предпочтительно от 20, 50, 100 или 200 до 800, 600, 500 или 400 мг в день, причем суточная доза может быть введена в виде разделенных доз от 1 до 4 раз в день, предпочтительно один или два раза в день; тогда как доза второго активного ингредиента находится в пределах от 1 до 100 мг, предпочтительно от 5, 10 или 20 до 80, 50 или 40 мг, причем дозу вводят с частотой в пределах от одной дозы каждые 7 дней до одной дозы ежедневно. Режим дозирования по этому воплощению, в частности, может быть применен, когда первый активный ингредиент доставляют посредством перорального введения или ингаляцией, а второй активный ингредиент вводят посредством подкожной инъекции. Подкожная инъекция второго активного ингредиента и режим дозирования по этому воплощению, в частности, могут быть использованы, когда второй активный ингредиент представляет собой этанерсепт.
Согласно настоящему изобретению дополнительно предложено применение фармацевтического продукта или набора по изобретению в производстве лекарства для лечения воспалительного расстройства.
Еще дополнительно согласно настоящему изобретению предложен способ лечения воспалительного расстройства, включающий одновременное, последовательное или раздельное введение:
а) (терапевтически эффективной) дозы первого активного ингредиента, который представляет собой антагонист Р2Х7 рецептора, причем антагонист Р2Х7 рецептора представляет собой производное адамантила; и
б) (терапевтически эффективной) дозы второго активного ингредиента, который представляет собой ингибитор фактора некроза опухоли (ФНО),
пациенту, нуждающемуся в этом.
В контексте настоящего описания термин "терапия" также включает "профилактику", если особо не указано обратное. Термин "терапевтический" и "терапевтически" следует истолковывать соответственно.
Профилактика, как ожидают, особенно необходима при лечении субъектов, которые страдали от предыдущего приступа или, как иначе считают, при повышенном риске данного состояния или расстройства. Субъекты с риском развития конкретного состояния или расстройства обычно включают тех, кто имеет это состояние или расстройство в семейном анамнезе, или тех, кто, как установлено посредством генетического тестирования или скрининга, является особенно чувствительным к развитию этого состояния или расстройства.
Изобретение также относится к тройной комбинированной терапии для лечения любого из следующих состояний: ревматоидный артрит, остеоартрит, остеопороз, псориаз, воспалительное заболевание кишечника, хроническая обструктивная болезнь легких (ХОБЛ), астма, аллергический ринит, или рак, или нейродегенеративные заболевания, такие как рассеянный склероз, болезнь Альцгеймера или удар.
Для лечения ревматоидного артрита фармацевтический продукт или набор по изобретению можно сочетать с "биологическими агентами", такими как антагонисты рецепторов IL-1 (например, анакинра) и рецептор-ловушка IL-1, рецептор IL-18, анти-IL-6 антитела, анти-СD20 антитела, анти-IL-15 антитела и CTLA4lg.
Подходящие агенты, используемые в сочетании с фармацевтическим продуктом или набором по изобретению, включают стандартные нестероидные противовоспалительные средства (в дальнейшем НПВС), такие как пироксикам, диклофенак, пропионовые кислоты, такие как напроксен, флубипрофен, фенопрофен, кетопрофен и ибупрофен, фенаматы, такие как мефенамовая кислота, индометацин, сулиндак, апазон, пиразолоны, такие как фенилбутазон, салицилаты, такие как аспирин. Ингибирующие циклооксигеназу доноры оксида азота (CINOD) и "модифицирующие заболевание агенты" (DMARD), такие как циклоспорин А, лефлуномид; циклесонид; гидроксихлорохин, d-пеницилламин, ауранофин или парентеральные или пероральные препараты золота также могут быть использованы.
Настоящее изобретение также относится к комбинации фармацевтического продукта или набора по изобретению с ингибитором биосинтеза лейкотриенов, ингибитором 5-липоксигеназы (5-LO) или антагонистом белка, активирующего 5-липоксигеназу (FLAP), выбранными из группы, состоящей из зилейтона; АВТ-761; фенлейтона; тепоксалина; Абботт-79175; Абботт-85761; N-(5-замещенных)-тиофен-2-алкилсульфонамидов; гидразонов 2,6-ди-трет-бутилфенола; метокситетрагидропиранов, таких как Зенека ZD-2138; соединения SB-210661; пиридинил-замещенных соединений 2n-цианонафталина, таких как L-739,010; соединений 2-цианохинолина, таких как L-746,530; соединений индола и хинолина, таких как МК-591, МК-886 и BAY×1005.
Настоящее изобретение также относится к комбинации фармацевтического продукта или набора по изобретению с рецепторным антагонистом лейкотриенов LTB4, LTC4, LTD4 и LTE4, выбранным из группы, состоящей из фенотиазин-3-онов, таких как L-651,392; амидино-соединений, таких как CGS-25019c; бензоксаламинов, таких как онтазоласт; бензолкарбоксимидамидов, таких как BIIL 284/260; и соединений, таких как зафирлукаст, аблукаст, монтелукаст, пранлукаст, верлукаст (МК-679), RG-12525, Ro-245913, иралукаст (CGP 45715А) и BAY×7195.
Настоящее изобретение также относится к комбинации фармацевтического продукта или набора по изобретению с ингибитором PDE4 (фосфодиэстеразы), включая ингибиторы изоформного PDE4D.
Настоящее изобретение также относится к комбинации фармацевтического продукта или набора по изобретению с антигистаминными антагонистами рецептора H1, включая цетиризин, лоратадин, дезлоратадин, фексофенадин, астемизол, азеластин и хлорфенирамин.
Настоящее изобретение также относится к комбинации фармацевтического продукта или набора по изобретению с гастропротекторным антагонистом рецепторов Н2 или ингибиторами протонного насоса (такими как омепразол).
Настоящее изобретение также относится к комбинации фармацевтического продукта или набора по изобретению с сосудосуживающим симпатомиметическим агентом агонистом 1- 2-адренорецепторов, включая пропилгекседрин, фенилэфрин, фенилпропаноламин, псевдоэфедрин, гидрохлорид нафазолина, гидрохлорид оксиметазолина, гидрохлорид тетрагидрозолина, гидрохлорид ксилометазолина и гидрохлорид этилнорэпинефрина.
Настоящее изобретение также относится к комбинации фармацевтического продукта или набора по изобретению с антихолинергическими агентами, включая бромид ипратропия; бромид тиотропия; бромид окситропия; пирензепин и телензепин.
Настоящее изобретение также относится к комбинации фармацевтического продукта или набора по изобретению с метилксантанинами, включая теофиллин и аминофиллин; кромогликатом натрия; антагонистом мускариновых рецепторов (М1, М2 и М3).
Настоящее изобретение также относится к комбинации фармацевтического продукта или набора по изобретению с модуляторами функции рецепторов хемокинов, таких как CCR1, CCR2, CCR2A, CCR2B, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9, CCR10 и CCR11 (для С-С семейства); CXCR1, CXCR3, CXCR4 и CXCR5 (для С-Х-С семейства) и СХ3СР1 для С-Х3-С семейства.
Настоящее изобретение также относится к комбинации фармацевтического продукта или набора по изобретению с миметиком инсулиноподобного фактора роста I типа (IGF-1).
Настоящее изобретение также относится к комбинации фармацевтического продукта или набора по изобретению с: (а) ингибиторами триптазы; (б) антагонистами фактора активации тромбоцитов (PAF); (в) ингибиторами интерлейкин-превращающего фермента (ICE); (г) ингибиторами IMPDH (инозинмонофосфатдегидрогеназы); (д) ингибиторами молекул адгезии, включая антагонисты VLA-4 (очень поздний антиген 4); (е) катепсинами; (ж) ингибиторами глюкозо-6-фосфат дегидрогеназы; (з) антагонистами кинин-В1 и В2-рецепторов; (и) противоподагрическими агентами, например колхицином; (к) ингибиторами ксантиноксидазы, например аллопуринолом; (л) урикозурическими агентами, например пробенецидом, сульфинпиразоном и бензбромароном; (м) агентами, усиливающими секрецию гормона роста; (н) трансформирующим фактором роста (TGF); (о) тромбоцитарным фактором роста (PDGF); (п) фактором роста фибробластов, например основным фактором роста фибробластов (bFGF); (p) колониестимулирующим фактором макрофагов-гранулоцитов (GM-CSF); (с) капсаициновым кремом; (т) антагонистами NK1 и NK3 рецепторов тахикинина, выбранными из группы, состоящей из NKP-608C; SB-233412 (талнетант); и D-4418; и (у) ингибиторами эластаз, выбранными из группы, состоящей из UT-77 и ZD-0892; (ф) ингибиторами индуцированной синтазы оксида азота (iNOS) или (х) хемоаттрактантной молекулой, гомологичной рецепторам, экспрессированной на клетках ТН2 (антагонисты CRTH2).
Фармацевтический продукт или набор по изобретению также может быть применен в сочетании с имеющимися терапевтическими агентами для лечения остеоартрита. Подходящие агенты, используемые в комбинации, включают стандартные нестероидные противовоспалительные средства (в дальнейшем НПВС), такие как пироксикам, диклофенак, пропионовые кислоты, такие как напроксен, флубипрофен, фенопрофен, кетопрофен и ибупрофен, фенаматы, такие как мефенамовая кислота, индометацин, сулиндак, апазон, пиразолоны, такие как фенилбутазон, салицилаты, такие как аспирин, ингибиторы индуцированной синтазы оксида азота (iNOS ингибиторы) и ингибирующие циклооксигеназу доноры оксида азота (CINOD), анальгетики (такие как парацетамол и трамадол), оберегающие хрящ агенты, такие как диацереин, доксицилин и глюкозамин и гиалуроновые кислоты, такие как гиалган и синвиск.
Фармацевтический продукт или набор по изобретению также может быть применен в сочетании с имеющимися терапевтическими агентами для лечения воспалительного заболевания кишечника (неспецифического язвенного колита и болезни Крона). Подходящие агенты, используемые в комбинации, включают 5-аминосалицилаты, тиопурины, азатиоприн и 6-мекапторурин.
Фармацевтический продукт или набор по изобретению также может быть применен в сочетании с противораковыми агентами, такими как эндостатин и ангиостатин, или цитотоксическими лекарственными средствами, такими как адриамицин, дауномицин, цис-платина, этопозид, таксол, таксотер и ингибиторы фарнезилтрансферазы, ингибиторы VegF (фактора роста сосудистого эндотелия), и антиметаболитами, такими как противоопухолевые агенты, особенно антимитотические лекарственные средства, включая алкалоиды барвинка, такие как винбластин и винкристин.
Фармацевтический продукт или набор по изобретению также может быть применен в сочетании с противовирусными агентами, такими как вирацепт, AZT, ацикловир и фамцикловир, и антисептическими соединениями, такими как валант.
Фармацевтический продукт или набор по изобретению также может быть применен в сочетании с блокаторами кальциевых каналов, агентами, понижающими уровень липидов, такими как фибраты, бета-блокаторами, Акф-ингибиторами (ингибиторами ангиотензин-конвертирующего фермента), антагонистами рецепторов ангиотензина-2 и ингибиторами агрегации тромбоцитов.
Фармацевтический продукт или набор по изобретению также может быть применен в сочетании с агентами для ЦНС, такими как антидепрессанты (такие как сертралин), противопаркинсонические лекарственные средства (такие как депренил, Л-допа, реквип, мирапекс, ингибиторы МАОВ (моноаминоксидазы В), такие как селегин и расагилин, ингибиторы КОМТ (катехол-O-метилтрансферазы), такие как тасмар, ингибиторы А-2, ингибиторы обратного захвата дофамина, антагонисты NMDA (N-метил-D-аспарагиновой кислоты), агонисты никотина, агонисты дофамина и ингибиторы нейрональной синтазы оксида азота) и лекарственные средства против болезни Альцгеймера, такие какдонепезил, такрин, пропентофиллин или метрифонат.
Фармацевтический продукт или набор по изобретению также может быть применен в сочетании с антиостеопорозными агентами, такими как ролоксифен, дролоксифен, лазофоксифен или фосомакс, и иммунодепрессивные агенты, такие как FK-506, рапамицин, цикпоспорин и азатиоприн.
Настоящее изобретение будет теперь дополнительно истолковано посредством ссылки на следующие иллюстрирующие примеры.
В примерах использовали следующие антагонисты Р2Х7:
1. Гидрохлорид N-[2-метил-5-(9-окса-3,7-диазабицикло[3.3.1]нон-3-илкарбонил)фенил]-трицисто[3.3.1.13,7]декан-1-ацетамида
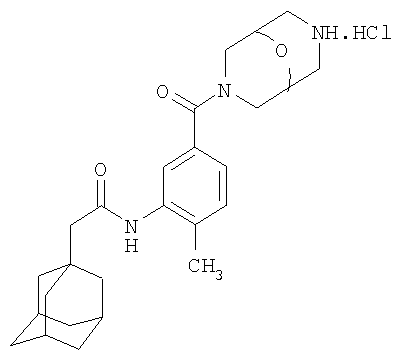
Антагонист Р2Х7 1 (гидрохлорид N-[2-метил-5-(9-окса-3,7-диазабицикло[3.3.1]нон-3-илкарбонил)фенил]-трицикло[3.3.1.13,7]декан-1-ацетамида) получали, как изложено ниже.
а) 3-(4-Метил-3-нитробензоил)-7-(фенилметил)-9-окса-3,7-диазабицикло[3.3.1]нонан
Оксалилхлорид (9,6 мл) в дихлорметане (30 мл) по каплям добавляли в течение 45 минут к охлажденному во льду раствору 4-метил-3-нитробензойной кислоты (10,0 г) в дихлорметане (320 мл), содержащему DMF (0,1 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, затем концентрировали под вакуумом. Хлорангидрид кислоты вводили в THF (320 мл) и охлаждали на ледяной бане перед добавлением N,N-диизопропилэтиламина (38 мл), затем порционно дигидрохлорида 3-(фенилметил)-9-окса-3,7-диазабицикло[3.3.1]нонана (16,0 г) (полученного, как описано в WO 01/028992). Реакционную смесь перемешивали в течение 18 часов, затем разбавляли этилацетатом (600 мл) и промывали водой (2×200 мл) и насыщенным бикарбонатом натрия (водн.) (3×150 мл), затем сушили (MgSO4), фильтровали и концентрировали с получением соединения, указанного в подзаголовке (18,5 г).
m/z=382.
б) 3-(3-Амино-4-метилбензоил)-7-(фенилметил)-9-окса-3,7-диазабицикло[3.3.1]нонан
Порошок восстановленного железа (7,9 г) добавляли в течение 15 минут к перемешиваемому раствору продукта со стадии (а) (18,0 г) и хлорида аммония (7,5 г) в смеси этанол/вода (3:1; 320 мл) при 70°С. Реакционную смесь нагревали с обратным холодильником в течение 2 часов, затем фильтровали и концентрировали под вакуумом. Остаток переносили в этилацетат (400 мл), промывали водой (2×150 мл), затем органическую фазу сушили (MgSO4) и концентрировали под вакуумом с получением соединения, указанного в подзаголовке (14,5 г).
m/z=352.
в) N-[2-Метил-5-[[7-(фенилметил)-9-окса-3,7-диазабицикло[3.3.1]нон-3-ил]карбонил]фенил]-трицикло[3.3.1.13,7]декан-1-ацетамид
Получали посредством способа стадии (а), используя 1-адамантануксусную кислоту и продукт со стадии (б). Перекристаллизация (этилацетат) давала соединение, указанное в подзаголовке.
m/z=528.
г) Гидрохлорид N-[2-метил-5-(9-окса-3,7-диазабицикло[3.3.1]нон-3-илкарбон]фенил]- [3.3.1.13,7]декан-1-ацетамида
4М HCl в 1,4-диоксане (8 мл) добавляли к раствору продукта со стадии (в) (13,0 г) в этилацетате (300 мл). Полученный осадок отделяли посредством фильтрации, затем суспендировали в этаноле (300 мл) и добавляли 5% палладий на углероде (1,2 г). Реакционную смесь перемешивали под давлением водорода в 3 атмосферы (303975 Па) в течение 36 часов. Затем добавляли метанол в атмосфере азота, затем удаляли катализатор посредством фильтрации, и фильтрат концентрировали под вакуумом.
Перекристаллизация (изопропанол:метанол 25:1; 800 мл) давала соединение, указанное в заголовке (9,1 г).
m/z=438 (M+H)+.
 н (400 МГц, d6-DMSO, Me4Si, 90°C) 9.06 (1H, s), 7.64 (1H, s), 7.25 (1H, m), 7.19 (1H, m), 4.15 (2H, s), 3.96 (2H, d, J=14 Гц), 3.35-3.23 (6Н, m), 2.26 (3H, s), 2.14 (2H, s), 1.96 (3H, уширенный s), 1.69-1.62 (12H, m). н (400 МГц, d6-DMSO, Me4Si, 90°C) 9.06 (1H, s), 7.64 (1H, s), 7.25 (1H, m), 7.19 (1H, m), 4.15 (2H, s), 3.96 (2H, d, J=14 Гц), 3.35-3.23 (6Н, m), 2.26 (3H, s), 2.14 (2H, s), 1.96 (3H, уширенный s), 1.69-1.62 (12H, m).
Пример 1
Фармакологический анализ для определения действия комбинаций ингибитор ФНО/антагонист Р2Х7 (без добавления агониста Р2Х7)
Моноциты периферической крови человека получали из крови здоровых добровольцев, собираемой в пробирки для гематологических исследований с ЭДТА. Моноциты выделяли посредством серийного градиентного центрифугирования и промывали для получения чистой популяции клеток. Затем добавляли липополисахарид (LPS) к клеточной суспензии в культуре ткани и инкубировали ее в течение 4-12 часов при 37°С. Затем к клеткам добавляли ингибитор ФНО и/или антагонист Р2Х7 или носитель. После инкубирования образцы клеточных супернатантов переносили в 96-луночный планшет для последующих измерений цитокинов и медиаторов. Образование медиаторов воспаления измеряли в клеточных супернатантах посредством специфических твердофазных иммуноферментных (ELISA) анализов для цитокинов IL-1, IL-18, ФНО и для других медиаторов, включая PGE2 (простагландины Е2), NO и матриксные металлопротеиназы (ММР). Определяли уровни высвобождаемых медиаторов в присутствии только антагониста Р2Х7 рецептора, или в присутствии только ингибитора ФНО, или в присутствии комбинации антагониста Р2Х7 рецептора с ингибитором ФНО. Затем сравнивали действие только антагонистов/ингибитора ФНО и в сочетании. Статистически значимые уровни ингибиторной активности сочетаний антагонист Р2Х7/ингибитор ФНО в отношении отдельного медиатора (IL-1 или ФНО) или различных медиаторов в сравнении с активностью, достигаемой при помощи либо только антагониста Р2Х7, либо ингибитора ФНО, является показателем повышенной эффективности при лечении заболевания.
Пример 2
Фармакологический анализ для определения действия комбинаций ингибитор ФНО/антагонист Р2Х7 (с добавлением агониста Р2Х7)
Моноциты периферической крови человека получали из крови здоровых добровольцев, собираемой в пробирки для гематологических исследований с ЭДТА. Моноциты выделяли посредством серийного градиентного центрифугирования и промывали для получения чистой популяции клеток. Затем добавляли липополисахарид (LPS) к клеточной суспензии в культуре ткани и инкубировали ее в течение 4-12 часов при 37°С. Затем добавляли исследуемые смеси с последующим добавлением BzATP агониста Р2Х7 рецептора. Исследуемые смеси могли содержать носитель в качестве контроля, антагонист Р2Х7 рецептора или комбинацию антагониста Р2Х7 рецептора с ингибитором ФНО. После инкубирования образцы клеточных супернатантов переносили в 96-луночный планшет для последующих измерений цитокинов и медиаторов. Образование медиаторов воспаления измеряли в клеточных супернатантах посредством специфических ELISA анализов для цитокинов IL-1, 1L-18, ФНО и для других медиаторов, включая PGE2, NO и матриксные металлопротеиназы (ММР). Определяли уровни высвобождаемых медиаторов в присутствии только антагониста Р2Х7 рецептора или в присутствии комбинации антагониста Р2Х7 рецептора с ингибитором ФНО. Затем сравнивали действие, производимое только антагонистом Р2Х7 и в сочетании с ингибитором ФНО. Статистически значимые уровни ингибиторной активности комбинаций антагонист Р2Х7/ингибитор ФНО в отношении отдельного медиатора (IL-1 или ФНО) или различных медиаторов в сравнении с активностью, достигаемой при помощи только антагониста Р2Х7, является показателем повышенной эффективности при лечении заболевания.
Пример 3
Оценка противовоспалительной активности комбинаций ингибитор ФНО/антагонист Р2Х7 в отношении артрита, вызванного клеточными стенками стрептококков, у крыс (Экспериментальная методика основана на методике, описанной Carlson R.P., Jacobsen P.В. "Comparison of adjuvant and streptococcal cell wall-induced arthritis in the rat" in Morgan D.W., Marshall LA., editors; In Vivo Models of Inflammation. Basel: Birkhauser Verlag; 1999.)
Артрит, вызванный клеточными стенками стрептококков (SCW), индуцировали в левом голеностопном суставе самок крыс линии Lewis.
Животных сенсибилизировали посредством внутрисуставной инъекции 5 мкг (в 20 мкл) SCW (Lee Laboratories) в левый голеностопный сустав. Отек голеностопного сустава оценивали через 3 дня после инъекции, а нечувствительных особей (животных без видимого отека голеностопного сустава) исключали из исследования. Чувствительных животных методом случайной выборки распределяли по исследуемым группам.
Артрит индуцировали через 21 день после сенсибилизации посредством внутривенной (в/в) инъекции SCW (100 мкг в 500 мкл физиологического раствора). Животных наблюдали и оценивали ежедневно вплоть до окончания через 6 дней после индукции. Крыс содержали на опилках и обеспечивали кормом и водой без ограничения.
В этом примере антагонист Р2Х71 давали перорально дозами по 30 мг/кг (4 мл/кг, дважды в день). Соединение давали дозами в виде суспензии в 1% (мас./об.) метилцеллюлозе в деионизированной воде и заново готовили каждый день. Дозирование начинали за 1 день до индукции артрита и продолжали вплоть до окончания на 6 день после индукции. Этанерсепт (0,5 мг/кг) вводили посредством подкожной инъекции (1 мл/кг) за 1 день до индукции артрита и затем на 1, 3 и 5 день после индукции.
Диаметры голеностопных суставов измеряли штангенциркулем ежедневно с 1 дня. Болевые пороги при механической стимуляции оценивали при использовании волосков фон Фрея на 1, 1, 3 и 5 день. Волоски применяли при увеличении нагрузки на область голеностопного сустава на подушечках обеих лап. Считали, что первый волосок, который вызывал реакцию отдергивания, служит болевым порогом.
Действия на отек голеностопного сустава и болевые пороги при механической стимуляции подсчитывали на основании площади под кривой (ППК) в виде суммы разностей от отдельных значений с 1 дня. Подсчитывали величину и направление взаимодействия и анализ данных проводили при помощи ANOVA с последующим использованием критерия Дуннетта на основе данных ППК (SAS, версия 8.01). Результаты суммировали в таблицу, приведенную ниже.
| |
% уменьшения ППК (по сравнению с контрольным возбудителем артрита) |
| Отек голеностопного сустава |
Болевой порог фон Фрея |
| Антагонист Р2Х71 |
28,5±13,5 |
21,1±10,9 |
| Этанерсепт |
44,0±4,7* |
7,4±6,8 |
| Антагонист Р2Х7 1 |
47,5±4,7* |
67,4±13,7** |
| + Этанерсепт |
|
Критерий взаимодействия р=0,085*** |
| *р<0,01, **р<0,001 по сравнению с контрольным значением возбудителя артрита, |
| *** показатель взаимодействия, свидетельствующий о большей, чем аддитивной, пользе при сочетании. |
Из вышеуказанных результатов можно видеть, что комбинация антагониста Р2Х7 1 и этанерсепта продемонстрировала положительное взаимодействие, производя значительно большее снижение болевого порога при механической стимуляции, чем можно было ожидать исходя из их отдельного применения. Обнаружение того, что два лекарственных средства обладают положительным действием на болевой порог фон Фрея в сочетании, которое не демонстрирует дополнительной пользы на отек голеностопного сустава, показывает, что сочетание лекарственных средств обладает сильным и неожиданно положительным действием на воспалительную боль суставов.
Формула изобретения
1. Фармацевтический продукт для лечения артрита, содержащий в сочетании препарат первого активного ингредиента, который представляет собой антагонист Р2Х7 рецептора, и препарат второго активного ингредиента, который представляет собой ингибитор фактора некроза опухоли (ФНО) этанерсепт, для одновременного, последовательного или раздельного применения в терапии, где антагонист Р2Х7-рецептора представляет собой соединение формулы (XI)
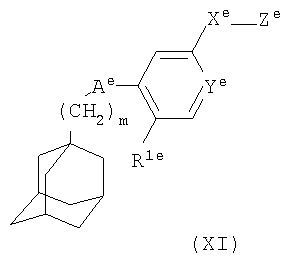 , ,
где m представляет собой 1, 2 или 3;
Аe представляет собой C(O)NH или NHC(O);
Ye представляет собой N или СН;
Xе представляет собой связь, СО, (СН2)1-6, О(CH2)1-6, (CH2)1-6NH(CH2)1-6, (CH2)1-6O(CH2)1-6, NH(CH2)1-6;
Ze представляет собой NR2eR3e;
R1e представляет собой галоген, циано, нитро, амино, гидроксил, C1-С6алкил или С3-С8циклоалкил, причем алкильная или циклоалкильная группа может быть возможно замещена одним или более чем одним атомом фтора;
R2e и R3e каждый независимо представляет собой атом водорода, C1-С6алкил или С3-С8циклоалкил, причем алкильная или циклоалкильная группа может быть возможно замещена одной или более чем одной группой, выбранной из гидроксила, галогена или C1-С6алкокси, или R2e и R3e вместе с атомом азота, к которому они присоединены, образуют 3-9-членное насыщенное моно- или бициклическое гетероциклическое кольцо, содержащее от 1 до 2 атомов азота и, возможно, атом кислорода, причем гетероциклическое кольцо может быть возможно замещено одной или более чем одной группой, выбранной из гидроксила, галогена или C1-С6алкокси;
или его фармацевтически приемлемые соль или сольват.
2. Композиция по п.1, где антагонист Р2Х7 рецептора представляет собой
2-хлор-5-[[2-(2-гидрокси-этиламино)-этиламино]-метил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-[3-[(3-гидроксипропил)амино]пропил]-N-(трицикло[3.3.1.1]дец-1-илметил)-бензамид,
(R)-2-хлор-5-[3-[(2-гидрокси-1-метилэтил)амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-[[2-[(2-гидроксиэтил)амино]этокси]метил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-[3-[3-(метиламино)пропокси]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)бензамид,
2-хлор-5-[3-(3-гидрокси-пропиламино)-пропокси]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-[2-(3-гидроксипропиламино)этиламино]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,2-хлор-5-пиперазин-1-илметил-N-(трицикло[3.3.1.1]дец-1-илметил)-бензамид,
2-хлор-5-(2,5-диазабицикло[2.2.1]гепт-2-илметил)-N-(трицикло[3.3.1.1]дец-1-илметил-)бензамид,
5-хлор-2-[3-[(3-гидроксипропил)амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамид,
5-хлор-2-[3-(этиламино)пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-пиридинкарбоксамид,
5-хлор-2-[3-[(2-гидроксиэтил)амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамид,
5-хлор-2-[3-[[(2S)-2-гидроксипропил]амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамид,
N-[2-метил-5-(9-окса-3,7-диазабипикло[3.3.1]нон-3-илкарбонил)фенил]-трицикло[3.3.1.13,7]дец-1-ацетамид
или фармацевтически приемлемые соль или сольват любого из них.
3. Набор для лечения артрита, содержащий препарат первого активного ингредиента, который представляет собой антагонист Р2Х7 рецептора, являющийся соединением формулы (XI), как определено в п.1, препарат второго активного ингредиента, который представляет собой ингибитор фактора некроза опухоли (ФНО) этанерсепт, и инструкции для одновременного, последовательного или раздельного введения препаратов нуждающемуся в этом пациенту.
4. Набор по п.3, где антагонист Р2Х7 рецептора представляет собой
2-хлор-5-[[2-(2-гидрокси-этиламино)-этиламино]-метил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-[3-[(3-гидроксипропил)амино]пропил]-N-(трицикло[3.3.1.1]дец-1-илметил)-бензамид,
(R)-2-хлор-5-[3-[(2-гидрокси-1-метилэтил)амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-[[2-[(2-гидроксиэтил)амино]этокси]метил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-[3-[3-(метиламино)пропокси]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-[3-(3-гидрокси-пропиламино)-пропокси]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-[2-(3-гидроксипропиламино)этиламино]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-бензамид,
2-хлор-5-пиперазин-1-илметил-N-(трицикло[3.3.1.1]дец-1-илметил)-бензамид,
2-хлор-5-(2,5-диазабицикло[2.2.1]гепт-2-илметил)-N-(трицикло[3.3.1,1]дец-1-илметил)-бензамид,
5-хлор-2-[3-[(3-гидроксипропил)амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамид,
5-хлор-2-[3-(этиламино)пропил]-N-(трицикло[3.3.1.13,7]дец-илметил)-4-пиридинкарбоксамид,
5-хлор-2-[3-[(2-гидроксиэтил)амино]пропил]-N-(трицикло [3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамид,
5-хлор-2-[3-[[(2S)-2-гидроксипропил]амино]пропил]-N-(трицикло[3.3.1.13,7]дец-1-илметил)-4-пиридинкарбоксамид,
N-[2-метил-5-(9-окса-3,7-диазабицикло[3.3.1]нон-3-илкарбонил)фенил]-трицикло[3.3.1.13,7] декан-1-ацетамид
или фармацевтически приемлемые соль или сольват любого из них.
5. Применение фармацевтического продукта или набора по любому из пп.1-4 в производстве лекарства для лечения артрита.
6. Применение по п.5,в производстве лекарства для лечения ревматоидного артрита.
7. Способ лечения артрита, включающий одновременное, последовательное или раздельное введение
а) (терапевтически эффективной) дозы первого активного ингредиента, который представляет собой антагонист Р2Х7 рецептора, являющийся соединением формулы (XI), как определено в п.1; и
б) (терапевтически эффективной) дозы второго активного ингредиента, который представляет собой ингибитор фактора некроза опухоли (ФНО) этанерсепт,
нуждающемуся в этом пациенту.
8. Способ по п.7 для лечения ревматоидного артрита. |
|
